An Algorithm to Predict the Possible SARS-CoV-2 Mutations
Abstract
An algorithm to determine the possible mutations that can occur in the S protein responsible of the Covid-19 in humans is designed. To do that, nine tridimensional sequences available in the Protein Data Bank similar to the initial strain sequenced in Wuhan (December 2019) are identified. The conditions driving this potential mutation are: (1) an accumulated number of mutations greater than (or equal to) 5 in each position; (2), a cumulative value of the different variations of Gibbs free energy less than -2.0 Kcal/mol; and (3), a squared fluctuation greater than 1.6 Å obtained according to calculations for normal mode analysis based on anisotropic network models (ANM) after averaging the first 20 vibration modes. The result is that 491 positions can mutate, while 424 positions did not provide any mutation. Finally, the results reveal that there are mutations that cannot be predicted, so more studies are needed to determine why they are present in the human population.
Author Contributions
Academic Editor: Amin Ataie, Babol university of Medical science, department of Pharmacology and toxicology, Iran.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2021 Raúl Isea
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
At the end of December 2019, the first episodes of Covid-19 were registered from patients from the Huanan Seafood Wholesale Market in the city of Wuhan (China) who presented a new atypical pneumonia, fever, cough, and in the most severe cases, dyspnea and bilateral lung infiltration. In view of this, on December 31, the Wuhan Municipal Health Commission reported the incident to the World Health Organization (WHO).
The genome of the virus was made public on January 2020 1. This study allowed the International Committee on Taxonomy of Viruses to rename it as SARS-CoV-2. From there, the virus began to spread to other cities in China, and later on to other countries in the world. In view of the high number of infections, the WHO determined a new Covid-19 pandemic on March 11.
The genome data was published in 2020 1. It was observed that it is a new betacoronavirus. It is 79% and 50% identical with respect to SARS-CoV and MERS-CoV, respectively. In other words, Covid-19 seems to be more related to the episode registered in 2002 rather than the incident that occurred in 2012. This observation is being investigated in more detail and will be published in a future work.
In this paper, there is a focus on the S protein because it is involved in the process of entering the virus into the receptor, and therefore it is a target for the design of possible vaccines against Covid-19. Recall that the S protein of SARS-CoV-2 consists of approximately 1,273 amino acids (aa), slightly higher than that found in SARS-CoV (1,255 aa).
Up to date, the emergency use of the vaccines Sputnik V from Russia, Sinopharm from China, among others, have been authorized. They are based on the analysis of the sequences that occurred in the initial studies of the virus, but a range of mutations can occur such that they are inefficient over time. Hence the need to predict possible mutations that can modify the effectiveness of vaccines 2, 3.
The first mutations registered in Brazil were found in a patient in Rio of Janeiro infected in October 2020 4. This mutation (E484S) spread to multiple countries including USA, Singapore, Argentina, Denmark, Ireland, England, Canada, etc.; but it was not found in Africa (remember that this variant is also known as 484 K.V2).
The second variant in Brazil has the following mutations: L18F, T20N, P26S, D138Y, R190S, K417T, N501Y, D614G, H655Y, and V1176F in the S protein 5, and was designated as B.1.1.248. It is interesting to see that the variant formed by K417T, E484K, and N501Y was independently named 28-AM-I, although both variants have also been assigned as P.1 variant as part of the B.1.1.28 nomenclature.
In view of this, nine three-dimensional (3D) structures of the S protein were selected to avoid any ambiguity in the results. Subsequently, an exploration of all the possible mutations that occur in each of the positions that make up the S protein was carried out, using the calculations of Gibbs free energy (ΔΔG) 6 as described in the next section. Finally, three scenarios will be handled in order to determine if a mutation in such a protein is possible.
Methodology
From one of the sequences published after the incident in the Wuhan market in December 2019, nine structural sequences of the S protein were selected from the Protein Data Bank (www.pdb.org), using the tools from the NCBI portal (blast.ncbi.nlm.nih.gov/). These sequences should provide a similarity higher than 98% with respect to the Wuhan sequence.
To consider a mutation as valid, two out of these three conditions should be met: (1) positions where at least five or more possible mutations (the half of the selected sequences) can occur; (2) amino acids that present a quadratic fluctuation equal to or greater than 1.6 Å obtained from an anisotropy network model calculations; and (3), the accumulation of the variation Gibbs energy is less than -2.0 Kcalc/mol.
The first mutations registered after the initial outbreak in Wuhan are analyzed, i.e.: Y28, A67, N74, W152, Y200, R273, F275, L276, E298, K300, T302, G485, A570, D614, A653, L752, P812, I818, G838, F1103, and V1104 7, 8, 9, 10, 11. In addition, to corroborate whether the two variants registered in Brazil (until February 2021) could be predicted with the results of the paper, the E484K mutation was initially registered in a patient infected in Rio of Janeiro in October 2020 3, and later a pool of mutations that occurred in the second Brazilian variant (also known as P.1) are present, which are: L18F, T20N, P26S. D138Y, R190S, K417T, N501Y, D614G, H655Y, and V1176F 3.
Results
The amino acid sequence of the S protein selected in the work corresponds to one of the first episodes registered in Wuhan (China) in December 2019, whose NCBI ID was MN908947. Thus, nine of their sequences were randomly selected from the result obtained with the Blastp program, which are deposited in the Protein Data Bank (PDB).
The nine sequences selected in humans correspond to the following PDB identifiers: 7JJI, 6VSB, 7KDI, 7KDJ, 6ZOW, 6XCM, 7CWL, 7K8S, and 7C2L. The next step was to calculate for each one of them, all the possible mutations that can occur from the calculation of the variation of Gibbs free energy after using the PoPMuSiC program.
(Figure 1) shows the number of mutations, the degree of exposure to the solvent, and the quadratic fluctuation in the region between Q14 and S98 in the 7CWL sequence. The figure shows how the number of mutations, the degree of exposure to the solvent, and the quadratic fluctuation are correlated throughout this region.
Figure 1.Selected region of the 7CWL sequence between Q14 until S98 of the S protein. The degree of exposure to the solvent is depicted in green color, the fluctuation due to the square displacement is depicted in light blue, and the number of mutations in red color.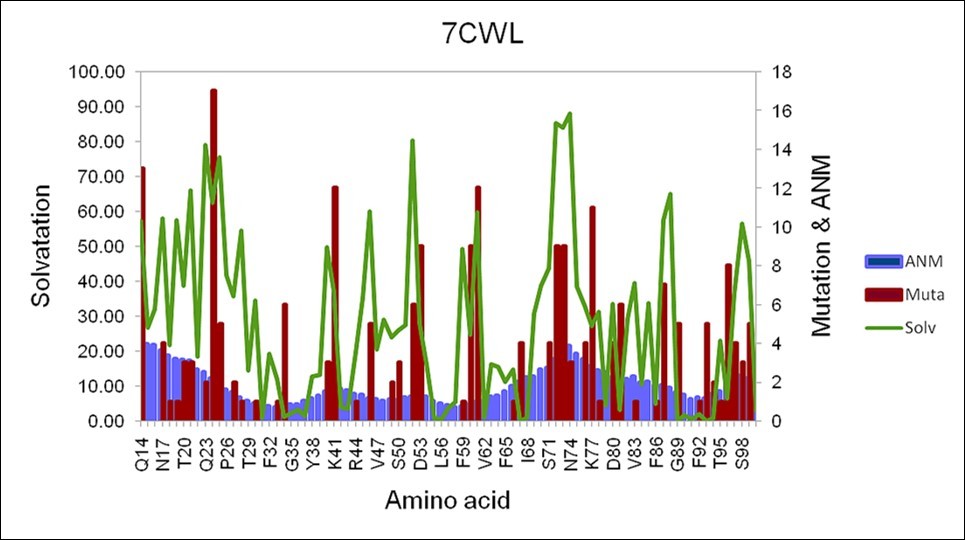
(Figure 2) shows the result of the evaluation of the possible mutations that may occur at position 200 of the S protein found in seven sequences selected in the work. This figure verifies the excellent agreement of the results obtained between all of them, as well as the average value depicted in blue (noted as <>).
Figure 2.Different results for the variation of the Gibbs free energy at position 200 of the S protein obtained in seven different sequences are shown. The average value is depicted in blue.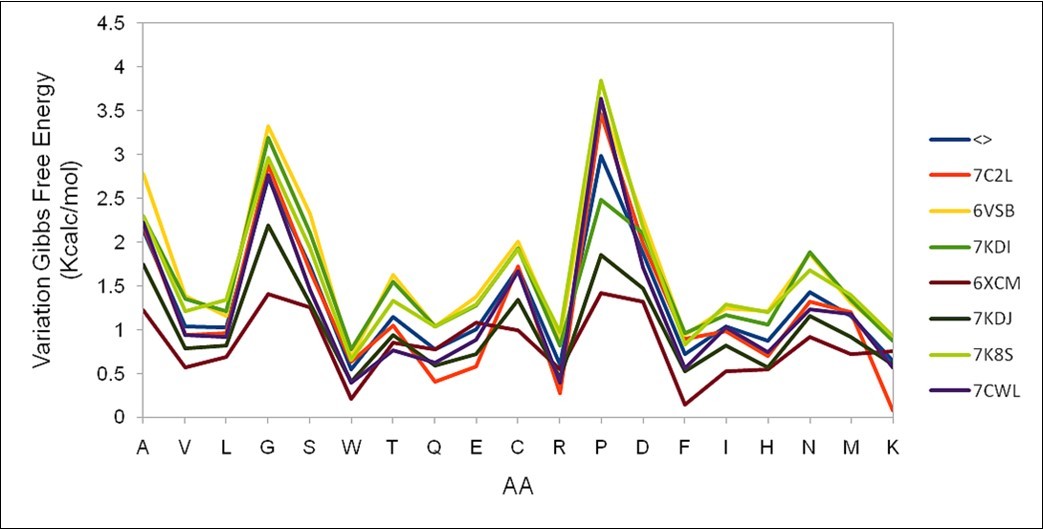
(Figure 3) shows the possible mutations in a small region between the positions Q14 and I68 in five different sequences. The distribution is not uniform as in the previous case, so the accumulated value of the mutations is considered instead of the average value.
Figure 3.Cumulative number of mutations obtained in the region between Q14 and I68 of the S protein obtained from five sequences considered in the work.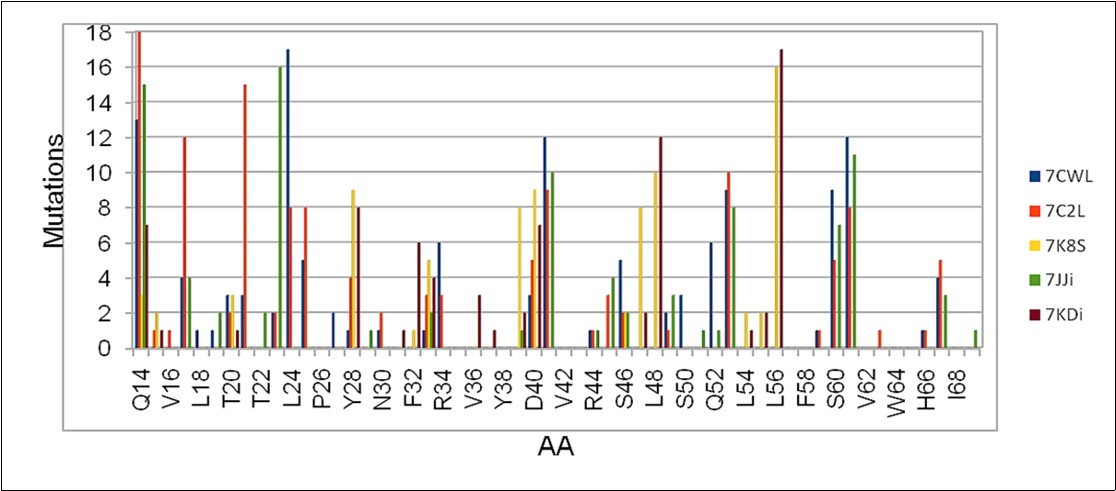
It should be noted that a total of 177,463 different Gibbs free energy calculations must be analyzed in the nine sequences selected in the work, so it was necessary to implement small scripts in the Python programming language to analyze those results.
The calculations revealed that 424 positions do not present any mutation in the nine sequences selected (indicated in Table 1). Table 2 shows the positions that meet two out of the three conditions proposed in this paper.
Table 1. The 424 amino acid positions where no mutation was detected:(Table 3) shows some of the mutations identified in the scientific literature, where the amino acid and position are indicated in the first column (AA) as well as the cumulative number of predicted mutations (Mut). The next column shows the accumulated value of the Gibbs free energy variations obtained by adding all the different contributions of the possible mutations (an example of this calculation is shown below), and the fourth column shows the mean square fluctuation obtained with an ANM methodology.
Table 3. Mutations observed in the S protein (AA) as well as the accumulated mutations obtained (Mut). The cumulative value of the different variations of Gibbs free energy at that position (ΔΔG accum), and the quadratic displacement obtained with ANM.| AA | Mut | ΔΔ G acumu . | <ANM> |
| Y28 | 14 | -1,58 | 1,8 |
| A67 | 16 | -2,96 | 3,2 |
| N74 | 14 | -2,29 | 4,7 |
| W152 | 1 | -0,04 | 4,8 |
| Y200 | 0 | 0,00 | 3,1 |
| R273 | 44 | -6,29 | 1,4 |
| F275 | 0 | 0,00 | 1,1 |
| L276 | 0 | 0,00 | 1,1 |
| E298 | 14 | -1,50 | 1,0 |
| K300 | 63 | -31,64 | 1,1 |
| T302 | 38 | -6,62 | 1,2 |
| G485 | 6 | -0,91 | 23,2 |
| A570 | 2 | -0,27 | 4,8 |
| D614 | 42 | -12,03 | 1,7 |
| A653 | 36 | -10,00 | 1,3 |
| L752 | 0 | 0,00 | 6,5 |
| P812 | 6 | -0,37 | 2,3 |
| I818 | 0 | 0,00 | 1,1 |
| G838 | 15 | -5,18 | 3,5 |
| F1103 | 0 | 0,00 | 3,4 |
| V1104 | 0 | 0,00 | 3,1 |
It is not possible to predict some mutations ie., Y200, F275, L276, L752, I818, F1103, and V1104 (Table 3). In fact, when checking the results in positions F275, L276, and I818, do not present any mutation. However, other mutations were predicted, such as Y28 or D614. The R273 and K300 positions are determined by a high number of mutations.
In order to understand the accumulated value of the Gibbs free energy variations (ΔΔG accum), the results obtained at position D614 are selected. This position has been found to be mutated to a GLY (G). The results obtained in each of the nine sequences are as follows:
6VSB: -1.04, CYS, PHE, GLY, HIS, ASN
6XCM: -2.11, CYS, PHE, GLY, HIS, ASN, TYR
7K8S: -1.35, CYS, GLY, HIS, ASN
7C2L: -0.58, CYS, GLY, HIS, ASN, VAL
7KDJ: GLY, 0,
7KDI: GLY, 0,
7JJI: -2.61, CYS, PHE, LEU, TRP, TYR
6ZOW: -1.48, CYS, PHE, GLY, HIS, ASN, PRO, THR, VAL, TYR
7CWL: -2.86, CYS, PHE, GLY, HIS, LEU, MET, ASN, TYR
Where the 6VSB sequence predicted five possible mutations (Cys, Phe, Gly, His, and Asn) such that the total sum of the five variations of the Gibbs free energy is equal to -1.04 Kcal/mol. The 7K8S sequence predicts four mutations and the cumulative Gibbs energy variation is -1.35 Kcalc/mol, and so on. Therefore, it can be verified that in position 614, 42 mutations present in 7 sequences are predicted, while two of them do not predict any mutation (7KDI and 7KDJ).
When reviewing the different mutations that are predicted in each of the seven sequences, it can be seen that the most frequent amino acid is a Gly (G), occurring 8 times. Cys (C) also appears 7 times, among others. Hence, an accumulated Gibbs energy of -12.03 Kcalc/mol was found (result of the sum of -1.04, -2.11, -1.35, - 0.58, -2.61, -1.48, and -2.86).
Finally, it is verified that the mutations registered in the new Brazil variants also appear in the results of this work, which are T20N, D138Y, R190S, K417T, N501Y, D614G, and H655Y. The L18 and P26 positions do not count on predicted mutations, and unfortunately the position V1176 was not present in the sequences.
Conclusion
This work determines the different positions where a mutation can occur in the S protein in order to explain the different variants that are occurring in SARS-CoV-2. It is interesting to note that it is possible to actually predict those observed in the new variant of Brazil, but it was not possible to explain some of the mutations detected at the beginning of the contagion by Covid-19 (L18, P26, Y200, F275, L276, L752 , I818, F1103 and V1104).
Acknowledgment
I’d like to acknowledge Rafael Mayo-Garcia for his comments on this manuscript.
References
- 1.Wu F, Zhao S, Yu B, Y M Chen, Wang W et al. (2020) A new coronavirus associated with human respiratory disease in China. , Nature 579(7798), 265-269.
- 2.Fontanet A, Autran B, Lina B, M P Kieny, Karim S S A et al. (2021) . , Lancet. Vol 397(10278), 952-954.
- 3.Santos dos, G W. (2021) Impact of virus genetic variability and host immunity for the success of COVID-19 vaccines. , Biomed Pharmacother 136, 111272.
- 4.C M Voloch, R da Silva F Jr, Almeida L G P de. (2020) Genomic characterization of a novel SARS-CoV-2 lineage from Rio de Janeiro,Brazil MedRxiv.
- 5.K N Harvey, P W Bird, J W-T Tang. (2021) Introduction of Brazilian SARS-CoV-2 484K.V2 related variants into the UK. , J Infect.(2021Feb3) 0163-4453.
- 6.Dehouck Y, J M Kwasigroch, Gillis D, Rooman M. (2011) PoPMuSiC 2.0: a web server for the estimation of protein stability of protein changes upon mutation and sequence optimality. , BMC Bioinformatics 12, 151.
- 7.Durmaz B, Abdulmajed O, Durmaz R. (2020) . Mutations Observed in the SARS-CoV-2 Spike Glycoprotein and Their Effects in the Interaction of Virus with ACE-2 Receptor. Medeni Med J 35(3), 253-260.
- 8.G B Chand, Banerjee A, G K Azada. (2020) Identification of twenty-five mutations in surface glycoprotein (Spike) of SARS-CoV-2 among Indian isolates and their impact on protein dynamics. Gene Rep. 21, 100891.
- 9.Chen R W J, Gao K, Hozumi Y, Yin C, Wei G-W. (2021) Analysis of SARS-CoV-2 mutations in the United States suggests presence of four substrains and novel variants. , Commun Biol 4, 228.
Cited by (2)
This article has been cited by 2 scholarly works according to:
Citing Articles:
Applied Mathematics and Nonlinear Sciences (2023) Crossref
Wen Yu - Applied Mathematics and Nonlinear Sciences (2022) Semantic Scholar
Applied Mathematics and Nonlinear Sciences (2022) OpenAlex