
Synthesis of N-Glycosyl Amides via Hydrolysis of Protected Glycosyl Oxazolines and Ritter-like Reactions of Native Carbohydrates
Abstract
A stereoselective synthesis of N-glycosyl amides was studied from available N-glycosyl oxazolines prepared by Ritter-like reactions of protected sugar acetonides. Hydrolysis reactions of the protected pentofuranosyl and hexafuranosyl oxazolines, as precursors of glycosyl amine derivatives, were carried out in the presence of silica gel in chloroform to giveN-α- and β-glycosyl amides in good yields after column chromatography on silica gel. Access to selectively blocked N-α-xylo-, -ribo-, β-arabino-furanosyl, α-glyco-, α-allo-furanosyl, α- and β-galactopyranosyl amides (twelve examples) useful for preparing modified N-glycosides was accomplished through a mild hydrolysis of sugar oxazolines with 2-alkyl substituents in acidic and neutral conditions. To further explore the scope of the BF3.Et2O-mediated approachdeveloped for N-furanosyl oxazolines, a stereoselective synthesis of protected N-α-hexopyranosyl oxazoline was fulfilled in a high yield from d-galactopyranose diacetonide derivative. The Ritter-like promoted reaction between D-arabinose and benzonitrile afforded 2-phenyl-β-d-arabinofurano-(1,2-d)-2-oxazoline as the main product. In acetonitrile the BF3.Et2O-KHF2-assisted reactions of unprotected native sugars were found to result in the formation of mixtures of N-furanosyl and pyranosyl acetamides.
Author Contributions
Academic Editor: Anubha Bajaj, Consultant Histopathologist, A.B. Diagnostics, Delhi, India
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2025 Grigorii G. Sivets
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Chemical modification of carbohydrates by introduction of different functional groups at an anomeric carbon to form glycosides is essential for the understanding of their biological functions 1. N-functionalization of sugars leading to N-glycosides with enhanced stability towards hydrolytic enzymes play a remarkable role in the field of glycobiological studies 2. The development of sugar mimetics with the C-N glycosidic linkage is interesting for medicinal chemistry, and designing effective therapeutic agents 3. The amide bond is an important connection found in natural compounds which can be used for attaching other biologically active molecules 4, 5 to carbohydrates. N-Glycosyl amides are stable under basic as well as acidic conditions and synthetic studies towards formation of the glycosyl amide linkage, approaches to structurally modified N-glycosides are of interest for carbohydrate chemistry.
N-Glycosyl amides and glycopeptides are known to take part in a variety of biological processes, and demonstrate a wide range of bioactivities, mainly because these types of carbohydrate derivatives can bind efficiently to specific sites of proteins, may be involved in numerous cell recognition processes 6, and can serve as glycomimetics. A series of glycosyl amides have been investigated as inhibitors of glycogen phosphorylase and the results of such studies provide evidence for interesting inhibitory properties of various N-β-glucopyranosyl amides (Fig.1, e.g., compounds I-II) 7, 8. N-Glycosyl amides as modulators of the activity of this enzyme may be used for developing a treatment for type 2 diabetes 8, 9. Furthermore, N-glycosyl amides III and IV were synthesized for evaluation of their potential antibacterial activities 10, 11. N-β- and α-fucosyl amides were identified as high-affinity ligands for lectin LecB 10 and it has also been reported that galacto-furanosyl or -pyranosyl amides may act as inhibitors of galactosidases or galactofuranosyltransferases 11, 12. Recently, N-sulfonyl amide derivatives of galaltopyranose V and glycopyranose VI as inhibitors of carbonic anhydrase II have been shown to exhibit antiglaucoma activities 13. Protected 2-chloro-1-acetamido sugar derivatives with gluco, galacto configuration (e.g., compound VII a) prepared from glycals and free amines (compound VII b) were found to be potently cytotoxic against the U-87 malignant glyoma (a brain tumor) cell line with IC-50 = 1 nm −22 ϻM 14. The glycoconjugate of the galactose with quinolinic acid derivative VIII involving amido linkagebetween sugar and a quinoline moiety exhibits cytotoxicity against cancer cells at the micromolar level 15 (Figure 1). In this context, it is worth noting interesting biological properties of bicyclic compound assigning to tetrahydropyrimidinone derivatives (6M3NP, the full title in ref.16) with an aromatic fragment and amide bond in pyrimidone moiety, and a new catalytic approach for its production. The compound showed antimicrobial and antioxidant activities. The anticancer activity of a phenyl acrylamide urea derivative was witnessed against variouscell lines such as MCF7, MDA-MB-231, and T47D (breast cancer) 16.
Figure 1.Biologically active N-glycosyl amides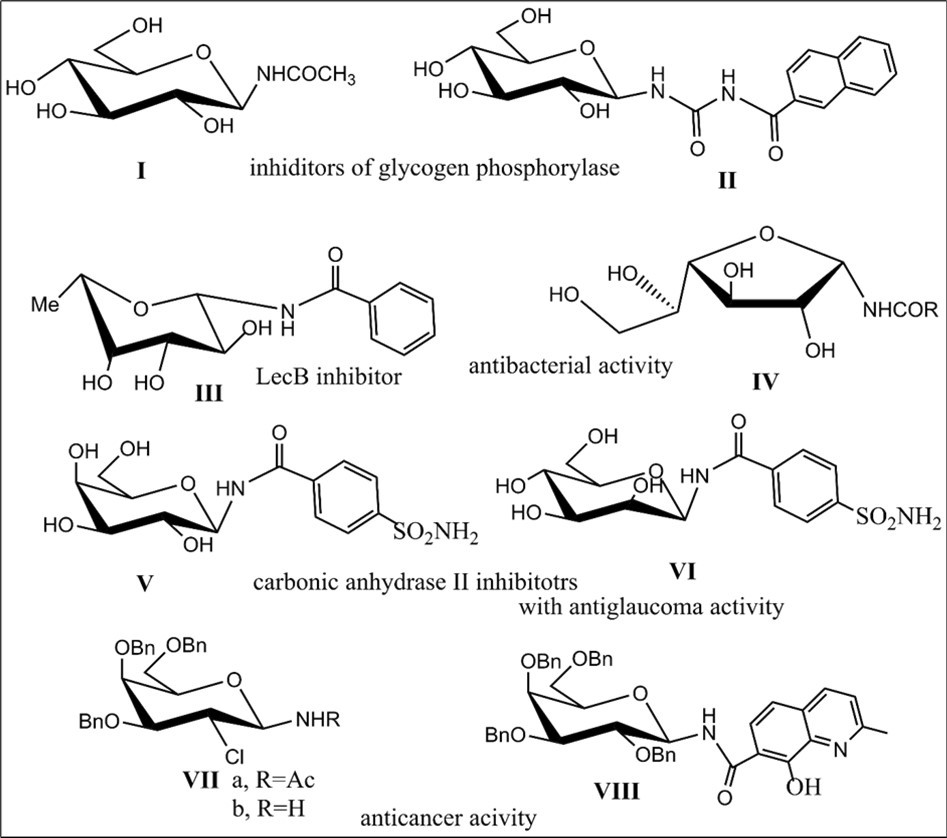
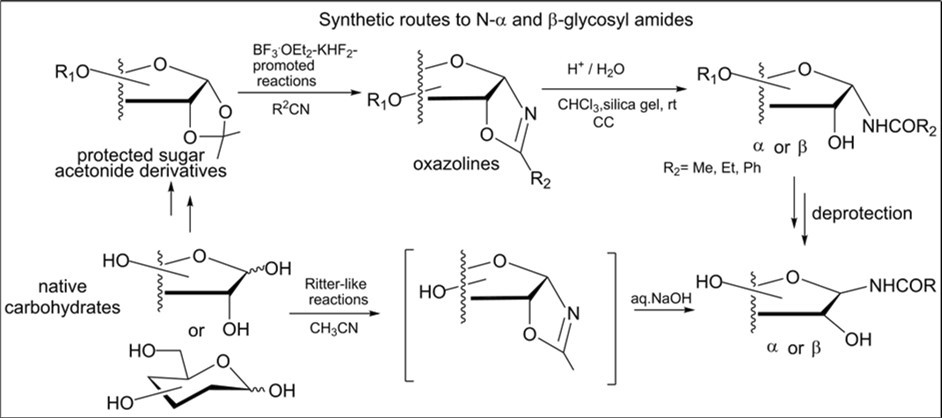
The obvious potential of N-glycosylamides in medicinal chemistryhas driven the search for stereoselective and efficient routes to produce these monosaccharide derivatives. Various approaches to access modified N-glycosides in different carbohydrate derivatives have been developed 1, 2, 4.Stereoselective synthetic methods to N-α- and β-glycosyl amides were earlier studied from protected and native carbohydrate precursors, and the most known of them are summarized in Scheme 1. The formation of N-glycosyl amides can be realized by preparation of intermediate aminosugars from blocked or unprotected sugars followed by their acylation with the acylating agent or via direct coupling of amides to glycosyl halides 2, 12, 17. The main drawback of a simple approach is the anomerization of glycosyl amines which leads to a mixture β- and α-amides after the acylation reaction.A facile and efficient synthesis to various β-glycosyl amines and amides was recently described from benzoylated α-glycosyl bromides by the ammonolysis reaction with aqueous ammonia 18. The method for preparing β- or a mixture of β-and α-glycosyl amides has been reported by decarboxylative reaction of unprotected sugar oximes with α-ketoacids in dimethylsulfoxide 19. Synthesis of protected β-glycosyl acetamides was also developed by treating 2,4-dinitrobenzenesulfonyl β-glycosyl amides derived from from glycosyl azides with thioacetic acid and cesium carbonate 20. Protected N-glycosyl acetamides as a mixture of N-β- and α-glycosides were obtained via a Ritter-type reaction of peracetylated D-hexopyranoses with acetonitrile in the presence of boron trifluoride etherate at room temperature 21.
Scheme 1.The known synthetic routes to N-glycosyl amides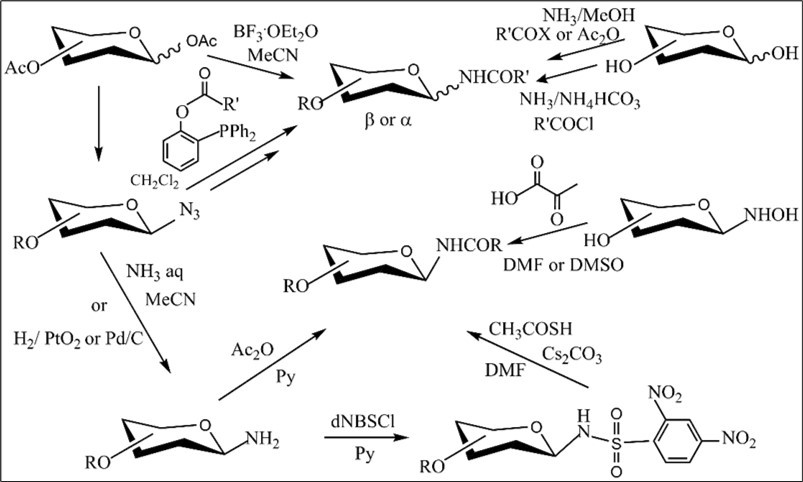
Glycosyl azides are used as valuable precursors for stereoselective synthesis of β-glycosyl amides because they possess chemical stability without anomerization at their anomeric centre and can be reduced by various methods prior to acylation reaction with the acyl derivatives. A common method, that is the most studied in carbohydrate chemistry, is based upon the Staudinger reactions including reduction-acylation process in which unprotected/blocked α- or β-glycosyl azide reacts with diphenyl phospanyl-phenyl esters 22, 23, 24. The diastereoselectivity of these reactions proves dependent on sugar protecting group and the configuration of the starting azide.
A limited number of synthetic routes was developed for the synthesis of α-glycosyl amides. Most of them include two steps and have been described for several hexo- and pentofuranose derivatives 11, 24. The method investigated by the Bernardi goup is founded on the traceless Staudinger ligation of various glycosyl azides of pyranose and furanose series with functionalized phosphines for stereoselective synthesis of α- or β-glycopyranosyl, α- or β-ribo- and arabinofuranosyl amides 23.
In extension of our previous study of the Ritter-like reaction in field of carbohydrates this paper reports exploration of synthetic routes to a series of novel and known N-glycosyl amides from available sugar oxazolines or native carbohydrates for further preparation and biological evaluation of modified N-glycosides and glycoconjugates with nucleosides.
Results and Discussion
Synthesis study of a series of N-glycosyl amides has been undertaken starting from a new approach developed for preparing N-glycosyl oxazolines 25 from the sugar acetonide derivatives. It was earlier shown that BF3.Et2O-mediated reactions of the 3,5-di-O-benzoylated d-xylofuranose-1,2-O-acetonide with nitriles followed by column chromatography on silica gel gave target 2-alkyl substituted oxazolinesalong with N-α-xylofuranosyl amides isolated in low yields. In the course of the present research, hydrolysis reactions of protected N-xylofuranosyl, ribofuranosyl and arabinofuranosyl oxazolines were studied on silica gel to prepare N-glycosyl amides.Formationof N-glycosyl amides from protected sugar oxazolines was found to proceed on silica gel under mild conditions in chloroform. Hydrolysis reactions of the protected oxazolines in the presence of silica gel gaveN-α- and β-glycosyl acetamides in good yields after column chromatography. Results on synthesis of a set of protected N-glycosyl amides are summarized in Table 1.
Table 1. Synthesis of selectively protected N-glycosyl amides via hydrolysis reactions of N-glycosyl oxazolines on silica gel| Entry | ProtectedD-furanosyl and pyranosyl oxazolines | Silica gel 60 H (70-230 mesh, Merck) | Time for hydrolysis reaction | N-glycosyl amide | (Yield, %) a |
| 1 |
1
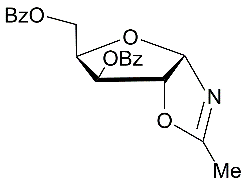
|
CHCl3 | 22 |
2
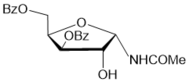
|
2 (84%) |
| 2 |
3
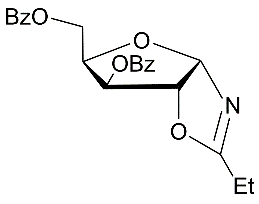
|
CHCl3 | 18 |
4
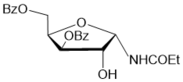
|
4 (85%) |
| 3 |
5
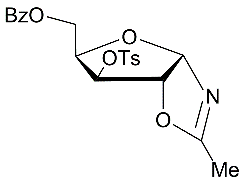
|
CHCl3 | 18 |
6
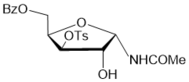
|
6 (85%) |
| 4 |
7
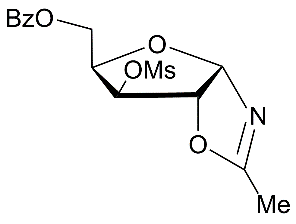
|
CHCl3 | 18 |
8
|
8 (86%) |
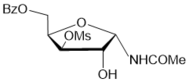
| 5 |
9
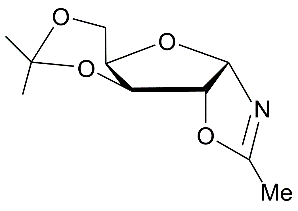
|
CHCl3 | 22 |
10
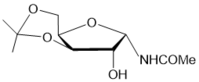
|
9 (78%) |
| 6 |
11
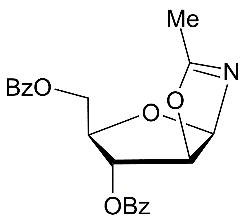
|
CHCl3 | 48 |
12
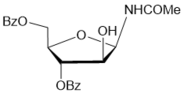
|
12 (84%) |
| 7 |
13
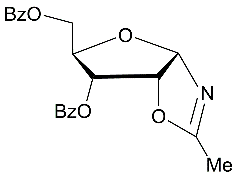
|
CHCl3 | 48 | 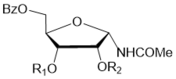 14,R1=Bz; R2=H15,R1=H; R2=Bz 14,R1=Bz; R2=H15,R1=H; R2=Bz |
14 (45%)and15 (11%) b |
|---|---|---|---|---|---|
| 8 |
16
|
CHCl3 | 22 |
17
|
17 (78%) |
| 9 |
18
|
CHCl3 | 18 |
19
|
19 (80%) |
| 10 |
20
|
CHCl3 | 18 |
21
|
21 (70%) |
| 11 |
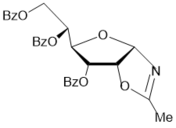
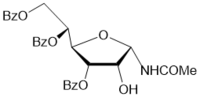
22
|
CHCl3 | 18 |
23
|
23 (78%) |
In the first place, with oxazolines in hands, syntheses of a series of selectively protected N-α-xylofuranosyl amides were investigated from the oxazolines under various conditions (Scheme 2). BF3.Et2O-mediated reactions of benzoylated d-xylofuranose 1,2-O-acetonide derivative with acetonitrile and propionitrile gave oxazolines in high yields after work-up of the reaction mixtures 25, but the formation of glycosyl amides was observed in 8-19% yields after chromatography using for elution mixtures of ethylacetate-petrolium ether. Partial hydrolysis of N-α-xylofuranosyl oxazolines took place during chromatography on silica gel. These findings may be attributable to moderate stability of the intermediate hemiorthoamidate derivatives forming after the nucleophilic addition of water to oxazolines 1 or 2 during chromatographic isolation, and their ability to undergo the regioselective cleavage into the α-glycosyl amides on silica gel. Further, the hydrolysis of oxazolines of xylo series was also studied under storing. Protected xylofuranosyl oxazoline 1 under a long storing (conditions a1) gave a mixture of products from which the benzoylated N-α-glycoside 4 was isolated in 67% yield. The oxazolines 2 and 3 (conditions a2-a3)also yielded N-α-xylofuranosyl acetamides 6-7 in67-86% yields after a long storing and chromatograpy or crystallization. Protected N-α-xylofuranosyl acetamide 10 was prepared from the 3,5-O-isopropylidene derivative of xylofuranosyl oxazoline 9 in a high 90% yield using mild conditions for the hydrolysis reaction (Scheme 2, conditions a4). We failed to carry out a direct synthesis of N-α-glycoside 10 by treatment of 9 with 73% aq. acetic acid at room temperaturedue toacidic hydrolysis ofthe oxazoline 9 is likely accompanied by the formation of acyclic by-products.
It was found thathydrolysis reactions of the protected N-α-xylofuranosyl oxazolines 1-4 and 9 proceeded on silica gel to giveselectively protected N-α-glycosyl acetamides in good yields (Scheme 2, Table 1, enties 1-5) after column chromatography on silica gel using chloroform and chloroform-methanol as eluents. Deacylation of N-α-xylofuranosyl amide derivatives 5 and 6 with cold saturated NH3/MeOH gave N-α-glycosides 11 and 12 in 77% and 81% yields, respectively (Scheme 2). Removing the isopropylidene protecting group in N-α-glycosyl amide 10 with 75% aq. CH3COOH furnished the target N-α-xylofuranosyl acetamide (11, 90%).
Scheme 2.Synthetic study of N-α-glycosyl amides with xylo configuration from D-xylose. Reagents and conditions: a1) 1, long storing at 5-8 0C, CC, 4, 67%; a2) 2, long storing at 5-8 0C, CC, 6, 86%; a3) 3, long storing at 5-8 0C, CC, 6, 60%; a4) 7, long storing at 5-8 0C, CC, 8, 90%; b1) 1, CHCl3, silica gel (entry1, table 1), 5, 84%; b2) 2, CHCl3, silica gel (entry 2, table 1), 6, 85%; b3) 3, CHCl3, silica gel (entry 3, table 1), 7, 85%; b4) 4, (entry 4, table 1), 8, 86%; b5) 9, CHCl3, silica gel (entry 5, table 1), 10, 78%; c) 9, 75% aq AcOH, rt, 20 h, CC, 11, 90%; d1) 5, NH3/MeOH, rt, 18 h, CC, 11, 77%; d2) 6, NH3/MeOH, rt, 18 h, CC, 12, 81%; f1) D-xylose CH3CN, KHF2, BF3.Et2O, rt, 4 h, CC, 11, 37%, 13, 5-6%; f2) D-xylose, EtCN, KHF2, BF3.Et2O, rt, 4 h, CC, 12, 28%; g1) 5, BzCl/Py, rt, 14, 42%, 15, 42%; g2) a mixture of 11 and 13, BzCl/Py, Et3N, rt, 14 (15%), and 15/16, 70%.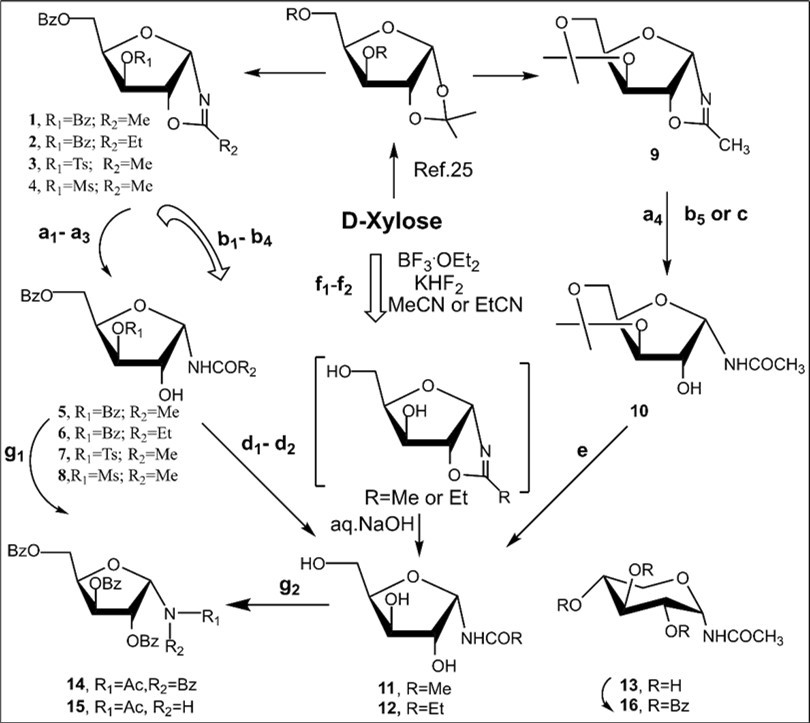
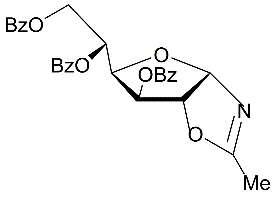
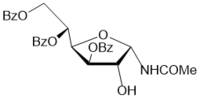
The Ritter-like BF3.Et2O-promoted reaction of d-xylose in acetonitrile afforded N-α-xylofuranosyl acetamide 11 as the main product in 37% yield along with formation of N-α-xylopyranosyl acetamide 13 (about 5-6%)and acyclic glycosyl amides as by-products (Scheme 2, conditions f1). The spectral data of N-α-xylofuranosyl acetamide prepared by the one-pot synthesis from D-xylose were identical to those of N-α-glycosуl acetamide 11 synthesized via protected N-α-xylofuranosyl acetamide 5 in five steps. However, efforts to separate N-α-glycosides 11 and 13 by column chromatograpy on silica gel were unsuccessful. Benzoylation of selectively protected N-acetyl d-xylofuranosyl amide 5 with benzoyl chloride in pyridine followed by column chromatography afforded individual perbenzoylated N-α-D-xylofuranosyl acetamide 14 (42%) and tri-O-benzoylated derivative 15 (42%). Treatment of a mixture of isomeric N-glycosides 11 and 13 (a separate fraction isolated by column chromatography on silica gel after the Ritter-like reaction), with benzoyl chloride in pyridine in the presence of triethylamine gave individual tetra-O-benzoylated N-α-xylofuranosyl acetamide 14 (15%) and a mixture of tri-O-benzoylated N-glycosides 15 and 16 (70%)(a ratio 3:1 according to 1H and 13C NMR spectral data), which were inseparable by chromatography on silica gel.The formation of N-α-xylofuranosyl and pyranosyl acetamides may proceed via generation of corresponding intermediate oxazolines forming during Ritter-likereactions of furanose and pyranose forms of D-xylose with acetonitrile in the presence of the Lewis acid. The BF3.Et2O-mediated reaction of d-xylose in propionitrile at room temperature afforded N-propionyl-α-d-xylofuranosylamide (12) which was isolated in 28% yield after column chromatography on silica gel. The structure of the N-α-glycoside was supported by comparison of NMR spectral data with those of 12 prepared by the multi-step approach from D-xylose through 3,5-di-O-benzoylated N-α-xylofuranosyl oxazoline 2 (Scheme 2). The magnitudes of 3JH-1,H-2 vicinal couplings 26 for protected N-acetyl-α-d-xylofuaranosyl amides 8 (J1,2 = 3.5 Hz), 7 (J1,2 = 3.8 Hz), 6 (J1,2 = 4.1 Hz) and 5 (J1,2 = 4.2 Hz) confirm their α-anomeric configurations and the cis-arrangement of H-1 and H-2 protons in the furanose rings, resonance signals of H-1 protons being displayed as doublet of doublets (JNH,H-1= 9-10 Hz) for synthesized N-α-xylofuranosyl amides in their NMR spectra measured in CDCl3. Absorption bands of the amidebond were revealed in the range of 1680-1505 cm-1 in IR-spectra of N-α-xylofuranosylamide derivatives. Furthermore, the downfield chemical shifts for C-1 signals (80-84 ppm) were observed in a series of N-α-xylofuranosyl amides5-8and10in comparison with the anomeric carbons of N-α-xylofuranosyl oxazolines1-4 and 925,which appeared at 100-101 ppmin the 13C NMR spectra, indicating attachment of acetamide group at the anomeric carbon in synthesized N-glycosides deriving from oxazolines. Resonance signals of H-1 protons for deprotected N-xylofuranosyl amides 11 and 12 as well N-xylopyranosyl amide 13 displayed as doublets for synthesized N-α-glycosyl amides in their NMR spectra measured in D2O or CD3OD. The low value of 3JH-1,H-2 vicinal coupling for N-acetyl-α-d-xylopyranosyl amide 13 (J1,2 = 3.1 Hz) is in good accordance with α configuration 12 at the anomeric centre of known xylopyranosyl amide derivatives. Further, synthetic approaches to N-β-arabinofuranosyl amides were studied from D-arabinose using the BF3.Et2O-mediated reactions. The Ritter-like reaction of d-arabinose in acetonitrile in the presence KHF2 and BF3.Et2O afforded N-β-arabinofuranosyl acetamide (19) in 21% yield along with of N-β-arabinopyranosyl acetamide 20 (18%)which were separated by column chromatography on silica gel (Scheme 3). The spectral data of N-β-arabinofuranosyl acetamide 19 prepared by the one-pot synthesis from D-arabinose were identical to those of the same N-β-glycosуl acetamide obtained in six steps through the hydrolysis reaction of the benzoylated oxazoline 17 on silica gel (entry 6, Table 1) followed by the deacylation oftheprotected N-glycoside 18 with ammonia in methanol (Scheme 3, conditions b).
Scheme 3.Synthesis of N-β-D-glycosyl acetamides with arabino configuration from D-arabinose. Reagents and conditions: a) 17, CHCl3, silica gel (entry 6, table 1), 18, 84%; b) 18, NH3/MeOH, rt, 18 h, 19, 68%; c) D-arabinose, CH3CN, KHF2, BF3.Et2O, rt, 4 h 10 min, CC, 19, 21%, 20, 18%.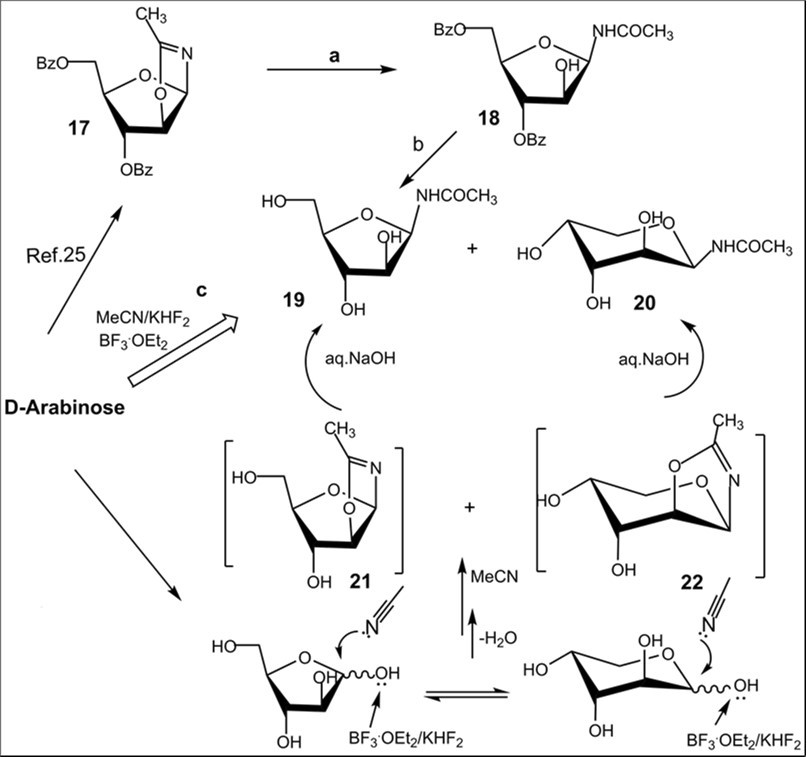
The structure ofN-β-arabinopyranoside 20 was supported by NMR spectral data and mass-spectroscopy. The formation ofisomeric N-β-glycosides 19 and 20 with acetamide group can be explained by generation of intermediate oxazolines (Scheme 3) during reactions of furanose and pyranose forms of D-arabinose in acetonitrile in the presence of BF3.Et2O 16 and KHF2 as promoters, and a subsequent stereoselective cleavage of oxazolines 21 and 22 under basic work-up of the reaction mixture.
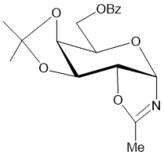
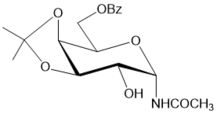
The BF3.Et2O-mediated reaction of d-arabinose in benzonitrile, unlike that of in acetonitrile, gave rise to 2-phenyl-β-d-arabinofurano-(1,2-d)-2-oxazoline (24) and 2-phenyl-β-d-arabinopyrano-(1,2-d)-2-oxazoline (25) at room temperature in 56% and 10% yields, respectively (Scheme 4, conditions b). After basic treatment of the reaction mixture isomeric 2-phenyl substituted glycosyl oxazolines were isolated by column chromatography on silica gel. The structure of 24 was supported by comparison ofits spectral data with those of 2-phenyl-β-d-arabinofurano-(1,2-d)-2-oxazoline prepared byremoving benzoyl protecting groups (NH3/MeOH) (Scheme 4, conditions a) in the oxazoline 23 ealier synthesized in six steps starting from D-arabinose 25. The assignment of structure for 2-phenyl-β-d-arabinopyrano-(1,2-d)-2-oxazoline 25 was made on the basis of its 1H and 13C NMR, mass spectral data. The benzoylated β-arabinofuranosyl oxazoline 23 as well as the unprotected oxazoline 24 with 2-phenyl substituent did not afford corresponding N-glycosyl amides under hydrolysis reactions in neutral or acidic conditions (on silica gel in chloroform), and they possess more stability in comparison with cis-fused benzoylated β-arabino- and α-xylofuranosyl oxazolines with 2-aliphatic alkyl substituents (Table 1, entries 1-4). Unlike 2-methyl-α-d-xylofurano-(1,2-d)-2-oxazoline and its derivatives, 2-phenyl-β-d-arabinofurano-(1,2-d)-2-oxazoline (24) was stable under a long storing, and formation of N-β-arabinofuranosyl amide 26 has not been found in the neutral conditions. Exploration of hydrolysis of oxazoline 24 was performed under various acidic conditions (aqueous trifluoroacetic acid, 6N aq. hydrocloric acid), but formation of N-glycosyl amide 26 was not observed due to toacidic hydrolysis ofthe oxazoline ring has been accompanied by the formation of acyclic by-products via cleavage of the furanose ring.
Scheme 4.Synthesis of 2-phenyl substituted N-β-D-glycosyl oxazolines of arabino configuration and acylated N-benzoyl-β-D-arabinofuranosylamide from d-arabinose. Reagents and conditions: a) 23, NH3/MeOH, rt, 18 h, 24, 74%; b) D-arabinose, PhCN, KHF2, BF3.Et2O, rt, 4 h 10 min, CC, 24, 56%, 25, 10%; c) 24, (iPr2SiCl)2O, Py, rt, 27, 98%; d) 27, CH2Cl2, aq. 33% HCl, 27, 88%; e) 24, 4ClBzCl, Py, rt, 29, 89%; f) 29, CF3SO3OH, CH2Cl2, rt, 31, 25%; g) 29, aq. HBF4, CH3CN, 00→ rt, 20 h, 20 h, 31, 19%, 32, 32%, recovery 23% of 29.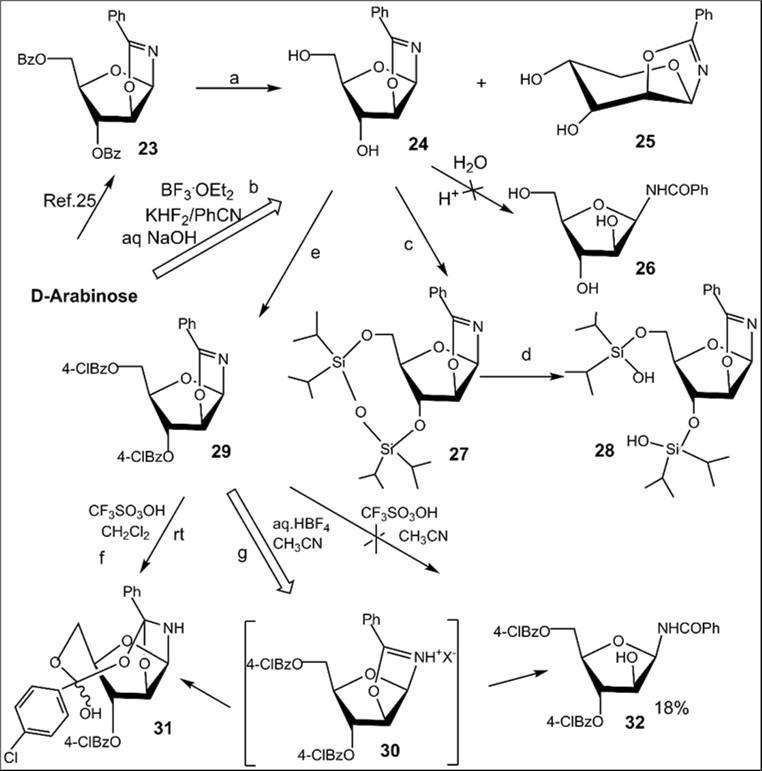
Further, to solve a challenge of a selective cleavage of 2-phenyl substituted glycosyl oxazolines, investigation of hydrolysis reactions of 3,5-di-O-protected 2-phenyl-β-d-arabinofurano-(1,2-d)-2-oxazolines was performed to prepare N-β-arabinofuranosyl benzamide derivatives (Scheme 4). Silylated derivative of oxazoline 27 was prepared by treatment of the oxazoline 24 with 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane in pyridine in 98% yield. The hydrolysis reaction of the oxazoline ring in 27 was studied under acidic conditions (aq.HCl/CH2Cl2) at room temperature. The cleavage of silyl protective group in 27 proceeded instead of selective transformations on 2-phenyl substituted oxazoline ring and silylated oxazoline derivative 28 was prepared in 88% yield. Benzoylation of the oxazoline 24 with 4-chlorobenzoyl chloride in pyridine at room temperature afforded 3,5-di-O-4-clorobenzoylated oxazoline 29 in 89% yield. The search for reaction conditions for selective hydrolysis of 2-phenyl substituted oxazoline ring in 3,5-di-O-acylated N-β-arabinofuranosyl oxazoline deivatives has been undertaken. Hydrolysis of benzoylated oxazoline 29 was studied in the presence 2-3 equiv. CF3SO3OH in acetonitrile, but no formation of protected N-β-arabinofuranosyl amide 32 was detected under tested conditions. Treatment of oxazoline 29 with 2-3 equiv. CF3SO3OH in CH2Cl2 at room temperature gave only adduct 31 in 25% yield. Conversions of by-product 31 was explored under mild acidic (on silica gel in chloroform) or basic conditions (aq. NaHCO3 in CH3CN/CH2Cl2), but no formation of target N-glycoside 32 was observed, only the starting arabinofuranose derivative was isolated in the both cases. Selective cleavage of the oxazoline ring in 29 was explored in the presence of a strong acid such as aqueous HBF4. The hydrolysis reaction of oxazoline 26 was investigated in acetonitrile with various access of aqueous HBF4. It was found that cleavage of oxazoline ring in 26 took place in the presence of a small access of the acid promoter to afford a mixture of products after column chromatography. N-β-arabinofuranosyl amide 32 (32%) was isolated after the hydrolysis reaction of the oxazoline under tested conditions followed by column chromatography on silica gel. It should be noted that cleavage of the oxazoline ring in 29 also results information of by-product 31 (14% yield) likely through coparticipation of 5-O-4-chlorobenzoyl group in the oxazolinium intermediate 30,and presumably 1-β-amino arabinose derivative (according to NMR data) as a result of acid-catalyzed hydrolysis of the 1,2-oxazoline ring of sugar in the presence of aqueous acid 27.1H NMR analysis of the reaction mixture after the mild basic treatment showed absence of N-β-glycosyl amide 32, but the formation of the latter along with by-sugar derivaives was found to take place during chromatography on silica gel likely through transformations of the oxazolinium intermediate. The structures of synthesized N-β-arabinofuranosyl benzamide derivative 32 as well by-product 31 were supported by 1H, 13C NMR and mass spectra.
Next, synthesis of protected N-ribofuranosyl acetamide derivatives was studied starting from D-ribose (Scheme 5). Hydrolysis reaction of the acylated α-d-ribofuranosyl oxazoline 33,prepared ealier fromD-riboseby Ritter-like reaction ofbenzoylated 1,2-O-isopropylidene-d-ribofuranose derivative 25,gavein neutral conditions (a long storing at 5-8 0C) 3,5-di-O-benzoylated N-α-ribofuranosyl acetamide 37 (70% of conversion of the oxazoline to the N-α-glycoside was determined according to 1H NMR data of a mixture of products), which was isolated in 62% yield after column chromatography on silica gel.
O-Deprotection of N-α-riboside derivative 37 with cold saturated NH3/MeOH at room temperature gave N-acetyl-α-d-ribofuranosyl amide (39)in 72% yield. Protected N-α-ribofuranosyl acetamide 37 (45%) along with 2,5-di-O-benzoylated N-α-ribofuranosyl acetamide 38 (15%) was also obtained using the hydrolysis reaction of the benzoylated oxazoline 33 on silica gel (entry 7, Table 1). In this case a mixture of isomeric protected N-ribofuranosyl acetamides 37 and 38 was prepared as a result of the hydrolysis reaction accompanied by migration of the 3-O-benzoyl group which led to 2,5-di-O-benzoylN-α-ribofuranosyl acetamide 38 (Scheme 5).The plausible mechanismofthe studied reaction leading to selectively di-O-acylated N-α-ribofuranosides from the oxazoline 33 islikely to include the formation of the oxazolinium itermediate 34 andasubsequent generation intermediates 35 and 36, which afford isomeric benzoylatedN-α-ribofuranosyl acetamides during column chromatography on silica gel using chloroform, chloroform-methanol as eluents. The Ritter-like reaction of d-ribose promoted with BF3.Et2O in the presence of KHF2 in acetonitrile at room temperature afforded N-α-ribofuranosyl acetamide (39) which was isolated by column chromatography on silica gel in a low 9% yield (Scheme 5, conditions e). The formation of 39 is likely to proceed through intermediate unstable oxazoline 40 forming as a result of BF3.Et2O promoted reaction of D-ribose with acetonitrile. Main products of the Ritter-like reaction were acyclic D-ribose derivatives with acetamide group, but their structures have not been established.
Scheme 5.Synthesis of N-D-ribofuranosyl acetamide derivatives from D-ribose. Reagents and conditions: a) 33, a long storing at 5-8 0C, 37, 62%; b) 33, CHCl3, silica gel (entry 7, table 1), 37, 45% and 38, 15%; c) 37, NH3/MeOH, rt, 18 h, 39, 72%; d) D-ribose, CH3CN, KHF2, BF3.Et2O, 00C→rt, 3 h, 39, 9%; e) peracylated d-ribofuranose 41, CH3CN, KHF2, BF3.Et2O, 00C→rt, 2 h, 1N aq. NaOH, 42α, 10%, 43β, 10%.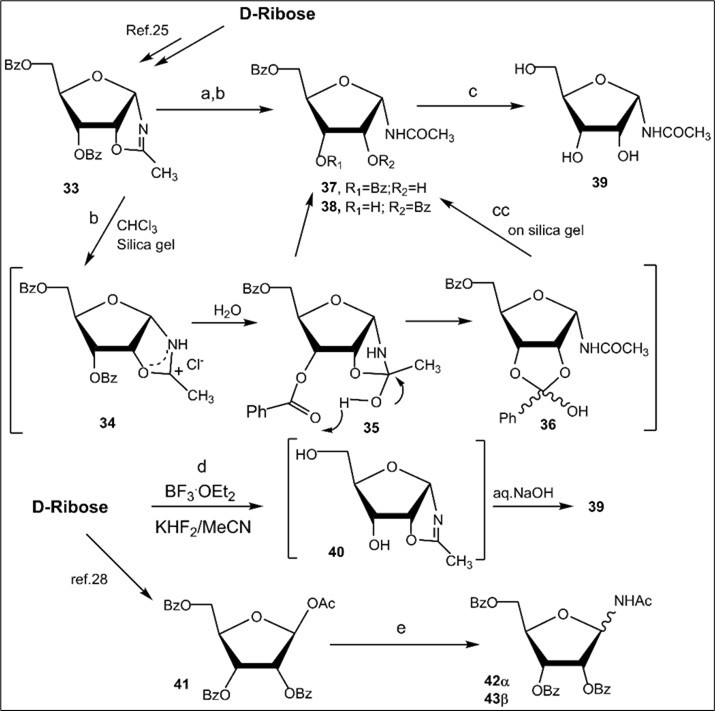
Further, the Ritter-like reaction of peracylated d-ribofuranose 41,prepared from d-ribose according to the known method 28, was studied in acetonitrile. Treatment of fully O-acylated d-ribose derivative 41 with KHF2-BF3.Et2O in CH3CN (conditions b3), unlike 1,3,5-tri-O-benzoyl-α-d-ribofuranose 25, afforded a mixture of products from which individual benzoylated N-α- and β-ribofuranosylacetamides 42α (10%)and 43β (10%)were isolated by column chromatography on silica gel. In this case the oxazoline was not obtained by the Ritter reactions in acetonitrile in the presence of a strong Lewis acid such as BF3.OEt2. Notably, in previous study on synthesis of nucleosides described earlier in the work29, the only benzoylated N-β-ribofuranosyl acetamide 42β has been prepared in 44% yield after the reaction of acetate 41 with acetonitrile under refluxing in the presence of trimethylsilyl triflate as a mild catalyst. Besides, Song and Hollingsworth 21 have reported a stereoselective synthesis of N-β-glycosyl amides from peracylated d-monosaccharides with gluco-, galacto- and manno-configurations using Ritter-type reactions with acetonitrile in the presence of methanesulfonic acid or BF3.Et2O.The stereoselectivity of studied anomeric Ritter-like reactions of d-glucose and mannose peracetates was explained by the anomerization via open-chain intermediates forming under acidic conditions.
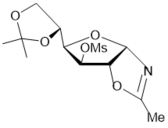
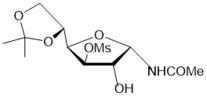
Exploring synthetic routes to N-hexofuranosyl amides, the hydrolysis reactions of protected N-α-glucofuranosyl and -allofuranosyl oxazolines prepared from D-glucose 25 were tested under conditions similar to N-xylofuranosyl oxazolines (Scheme 3). Hydrolysis of benzoylated N-α-glucofuranosyl oxazoline 44 as well as benzoylatedN-α-xylofuranosyl oxazoline 1 proceeded under storing in the presence of traces of water (Scheme 6, conditions a) to afford the N-α-glucofuranosyl acetamide derivative 45 (80% of conversion into the N-glycoside according to 1H NMR data), which was isolated in 66% yield after chromatography on silica gel.
The hydrolysis reaction of the benzoylated oxazoline 44 on silica gel in chloroform (entry 6, Table 1) gave N-α-glucoside 45 in 58% yield after column chromatography. The deprotection of the latter with NH3/MeOH or 1 M MeONa in methanol, using the Zemplén-deacylation conditions, gave N-α-glucofuranosyl acetamide 46 in 65% and 72% yields, respectively. The benzoylated allofuranosyl oxazoline 47 prepared by Ritter-like reaction of protected 1,2-O-isopropylidene-α-D-allofuranose, which was synthesized in two steps from available D-glucose diacetonide according to the known method 30, affordedacylatedN-α-allofuranosyl acetamide 48 (80%)under stereoselective hydrolysis on silica gel (Scheme 6). After a long storing the oxazoline 47 gave3,5,6-tri-O-benzoylated N-α-allofuranosyl acetamide 48 in a high 85% yield (Scheme 6, conditions f). The Ritter-like reaction of d-glucose in acetonitrile at room temperature gave rise to N-α-d-glucofuranosyl acetamide (46) and isomeric α-d-glucopyranosyl acetamide (51) in 13% and 37% yields, respectively, which were isolated by column chromatography on silica gel.
Scheme 6.Synthesis of N-α-D-glycosyl acetamides of glyco and allo configuration from D-glucose. Reagents and conditions: a) 44, a long storing at 5-8 0C, 45, CC, 66%; b) 44, CHCl3, silica gel (entry 8, table 1), 45, 78%; c) 45, NH3/MeOH, rt, 18 h, 46; 65%; d) 45, 1 M MeONa/MeOH, rt, 14 h, 46, 72%; e) 47, (entry 9, table 1), 48, 80%; f) 47, a long storing at 5-8 0C, 48, 85%; g) D-glucose, CH3CN, KHF2, BF3.Et2O, rt, 4 h, CC, 46, 13%, 51, 37%.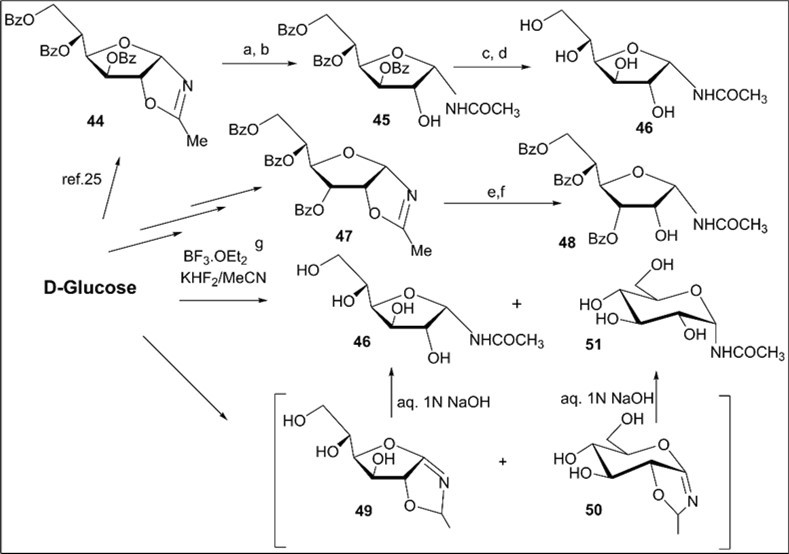
The formation of N-glycoside 46 was supported by comparison ofits spectral data with those of N-α-d-glucofuranosyl acetamide prepared byremoving benzoyl protecting groups (NH3/MeOH) in the acylated N-α-glucofuranosyl acetamide 45 ealier synthesized via the intermediate oxazoline 44. Besides, the structure of N-glycopyranoside 51 was confirmed byNMR spectral data as in the case of the preparation of N-α-d-glucopyranosyl acetamide described earlier via azidosugar 22. The most probable mechanistic pathway of the BF3.Et2O-mediated Ritter-like reaction leading to isomeric N-glyco-furanosyl and -pyranosyl acetamides includes the formation of intermediate 2-methyl-β-d-glucofurano-(1,2-d)-2-oxazoline (49) and 2-methyl-β-d-glucopyrano-(1,2-d)-2-oxazoline (50) from D-glucose in the presence the Lewis acid followed by hydrolysis reactions under basic work-up the reaction mixture (Scheme 6).
In extension of this study, synthesis of protected N-α-galactopyranosyl acetamides was investigated using benzoylated N-α-galactopyranosyl oxazoline 53 which was prepared by the BF3.Et2O-KHF2-promoted reaction of the 6-O-benzoyl d-galactopyranose1,2;3,4-diacetonide derivative 52 with acetonitrile in a high 93% yield without chromatography on silica gel (Scheme 7, conditions a). A selective hydrolysis of the protected N-α-galactopyranosyl oxazoline 53 proceeded on silica gel in chloroform to give the N-α-galactopyranosyl acetamide derivative 55 (78%) likely through the formation of the intermediate hemiorthoamidate derivative 54 in mild acidic conditions (Scheme 7, conditions b). Full O-deprotection of intermediate N-glycoside 55, isolated by column chromatography on silica gel, afforded the target N-α-galactopyranosyl acetamide 56 (58% yield over two steps, Scheme 7, conditions c).N-Acetyl-α-D-galactopyranosyl amine 56 has earlier been synthesized by acetylation of α-D-glycopyranosyl amine with acetic anhydride and studied along with a number of N-acyl-α-galactopyranosyl amines as potential competitive inhibitors of α-D-galactosidase 31. Compound 56 displayed inhibiting properties towards α-D-galactosidase from Trichoderma reesei. The Ritter-like reaction of d-galactose in acetonitrile at room temperature gave rise to N-α-d-galactofuranosyl acetamide (57) and isomeric α-d-galactopyranosyl acetamide (56) in 18% and 15% yields, respectively, after column chromatography on silica gel. Notably, as opposed to benzoylated N-α-pentofuranosyl oxazolines 1 and 33 or N-α-glucofuranosyl oxazoline 44 (Schemes 2, 5 and 6), the protected N-α-galactopyranosyl oxazoline 53 did not result in N-α-galactopyranosyl acetamide 55 under the mild conditions for the hydrolysis reaction (conditions e). In this case, after a long-term storing of the individual N-α-galactopyranosyl oxazoline 53 in the presence of traces of H2O, the formation of N-β-galactopyranosylamine derivative 59β was established from 1H and 13C NMR spectra taken in CDCl3 (82% yield, conditions e). The structure of 59β was confirmed from 2D NMR spectroscopy and mass spectrum (Experimental part). Hydrolysis reaction of the oxazoline 53 in the presence of aq. HCl in methylene chloride gave β-amine 59β in 34% yield according to 1H NMR spectrum of the reaction mixture (Scheme 7, conditions f).Besides, full O- and N-acetylation ofthe β-amine 59β (conditions g) gave the only 2-O-acetylated N-β-galactopyranosyl acetamide derivative 60 in 75% yield after chromatography on silica gel. The chemical shifts and large magnitudes of 3J1,2 vicinal couplings for H-1 protons observed in 1H NMR spectra of the β-amine 59β (4.03 ppm, d, J1,2 = 8.7 Hz) and the β-amide 60 (5.12 ppm, t, J1,2 = JH-1, NH = 9.3 Hz) are indicativeof the β-anomeric configuration of amino and acetamido groups at the anomeric centers. It is necessary to note that the values of H-1 coupling constantsfor 59βand60are in good accordance with those(8.3-9.8 Hz), which are characteristic for the known acylated N-β-galactosyl amides described earlier by Pleuss and Kunz 32.
Scheme 7.Synthesis of protected N-α-d-galactopyranosyl oxazoline and N-α- and β-d-galactosyl acetamides from D-galactose. Reagents and conditions: a) 52, CH3CN, KHF2, BF3.Et2O, rt, 18 h, 1N aq NaOH; 53, 93%; b) 53, CC on silica gel, (entry 11, table 1), 55, 78%; c) i) 55, 80% aq CH3COOH, 50-550C, 18 h, ii) NH3/MeOH, rt, 18 h, 56, 58% over two steps; d) D-galactose, CH3CN, KHF2, BF3.Et2O, rt, 4 h 10 min, CC, 56, 15%, 57, 18%; e) 53, a long-term storing at 5-8 0C, 82%, 59β; f) 53, CH2Cl2, aq.HCl, 59β, 34%; g) 59β, AcCl, CH2Cl2, Et3N, rt, 60, 75%.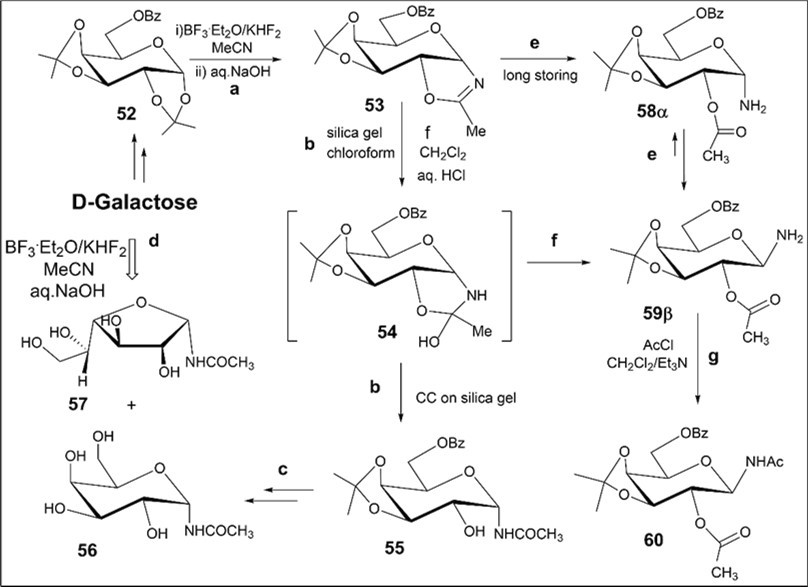
The formation of the N-β-galactosylamine derivative 59β arises from the anomerization 27, 33 of the intermediate α-glycosyl amine 58α which produces under a long-term storageof N-α-galactopyranosyl oxazoline 53. Selectively protected N-β-galactopyranosylamine 59β may be used for preparing N-sulfonyl amide derivatives of galactopyranose with antiglaucoma activity.
Thestructures of synthesized N-α- and β-galactosyl amides were confirmed by NMR and mass spectra. New synthetic approaches to N-glycosyl amine derivatives were studied from D-galactose via the Ritter-like reaction of d-galactopyranose1,2;3,4-diacetonide derivativefor preparation of intermediate N-α-D-galactopyranosyl oxazoline followed by hydrolysis reactions with the formation of N-α- and β-galactopyranosyl amines or α-amide. It was shown that the efficient method developed for N-furanosyl oxazolines 25 from sugar acetonides can be utilized for stereoselective synthesis of protected N-hexopyranosyl oxazoline from d-galactopyranose diacetonide derivative. A series of synthesized protected N-glycosyl amides as well as N-glycopyranosyl oxazoline(s) can be used as potential glycosyl donors in carbohydrate chemistry for the preparation of O- and N-glycosides32.
Conclusion
In summary, two synthetic routes to N-glycosyl amides were investigated from D-pentose and hexose sugars. Hydrolysis reactions of N-α-furanosyl oxazolines, prepared from protected D-pentofuranose and hexafuranose 1,2-O-acetonides, have been studied in neutral and acidic conditions to prepare N-α-furanosyl amides. Protected glycosyl 1,2-oxazolines with 2-aliphatic alkyl substituents were found to undergo gradual conversions to N-glycosyl amides in neutral conditions in the presence of traces of water. It has been shown that hydrolysis reactions of blocked 2-methyl substituted N-glycosyl oxazolines proceeded on silica gel in chloroform to give selectively protected N-α- or β-glycosyl amides in good yields. N-Glycosyl oxazolines with 2-phenyl substituent e.g., 3,5-di-O-benzoylated 2-phenyl-β-d-arabinofurano-(1,2-d)-2-oxazoline as well as the unprotected arabinofuranosyl oxazoline did not afford corresponding N-glycosyl amides in neutral or acidic conditions due to they possess more stability in comparison with cis-fused β-arabino- and α-xylofuranosyl oxazolines with 2-alkyl substituents.
Reaction conditions for selective cleavage of 2-phenyl substituted β-arabinofuranosyl oxazolines were explored and the hydrolysis of the protected oxazoline in the presence of HBF4 in acetonitrile gave rise to benzoylated β-arabinofuranosyl benzamide in a moderate yield after chromatography on silica gel. In addition, syntheses of N-α-gluco-, allofuranosyl, and N-α- or β-galactopyranosyl amides of biological interest were accomplished through a mild hydrolysis of protected N-α-glycosyl oxazolines. In the second direct approach, N-furanosyl and pyranosyl acetamides were obtained through the BF3.OEt2-KHF2-mediated reactions of a series of native sugars in acetonitrile at room temperature. The synthesized N-glycosyl amides will be used for preparation and future biological evaluation of modified N-glycosides and glycoconjugates with anticancer nucleosides.
Experimental part
General information
Column chromatography was performed on silica gel 60 H (70-230 mesh; Merck, Darmstadt, Germany), and thin-layer chromatography (TLC) on Merck silica gel aluminum 60 F254 precoated plates. All commercially available reagents were used without further purification. 1H and 13C NMR spectra were recorded in CDCl3 and CD3OD with a Bruker Avance-500-DRX spectrometer at 500.13 and 126.76, respectively. 1H and13C NMR chemical shifts (δ, ppm) are relative to internal chloroform peak (7.26 ppm for 1H and 77.0 for 13C NMR). Splitting patterns were reported as following: s: singlet, d: doublet, t: triplet, m: multiplet. J values are reported in Hz. Optical rotations were measured with Autopol III automatic polarimeter. IR spectra were measured with on PerkinElmer Spectrum 100FT-IR spectrometer. Melting points were determined on a Boetius apparatus and were uncorrected. High resolution mass spectra (HRMS) were recorded on an Agilent Q-TOF 6550 Instrument (USA) using ESI (electrospray ionization).
General procedure for preparation of protected N-glycosyl amides by hydrolysis reactions of N-glycosyl oxazolines on silica gel (Table 1).
The oxazoline (0.5 – 1.6mmol) prepared from corresponding protected D-pentofuranose or -hexafuranose acetonide was dissolved in chloroform and placed to the top of a silica gel column (60 H, 70-230 mesh; Merck, Darmstadt, Germany) prepared in chloroform, which was washed with a small volume of chloroform. After 18-48 h a silica gel column was washed with chloroform and further column chromatography gave N-glycosyl amides in 70-86% yields using mixtures of chloroform-methanol for gradual elution.
3,5-Di-О-benzoyl-N-acetyl-α-d-xylofuranosylamide(5)from the oxazoline 1:
a1. The N-xylofuranosyl oxazoline derivative 1 (0.04 g, 0.1 mmol) was kept at 5-8 oC for 6 weeks. The oily residue was chromatographed on a silica gel, using for elution chloroform, chloroform–methanol 10:1 and 5:1 to give (0.028 g, 67%) N-α-d xylofuranosylacetamide 5.
b1.The 3,5-di-O-benzoyl-protected oxazoline 1 (310 mg, 0.78 mmol) was dissolved in chloroform and placed to the top of a silica gel column which was prepared in chloroform. After 24 h at room temperature a silica gel column was washed chloroform and then further gradual elution with chloroform-methanol 30:1, 15:1 and 10:1 gave (0.275 g, 85%) of 3,5-di-О-benzoyl-N-acetyl-α-d-xylofuranosylamide (5) as oil.
(α)D20 +15.8 (c 1.0, CHCl3). IR (KBr): ν 3388, 1722, 1656, 1526, 1271, 1180, 1106 сm-1. 1Н NMR (500 MHz, CDCl3) δ = 7.51-7.99 (m, 10H, 2 x СОC6H5), 6.86 (d, 1Н, J = 9.0 Hz, NHCOMe), 6.07 (dd, 1Н, J1,2 = 4,2 Hz, Н-1), 5.52 (dd, 1Н, J3,4 = 4.0, J3,2 = 1.5 Hz, Н-3), 4.76-4.80 (m, 1Н, Н-4), 4.59 (dd, 1Н, Н-5), 4.57 (dd, 1Н, Н-5′), 4.37 (dd, 1Н, J2,1 = 4.2, J2,3 = 1.5 Hz, Н-2), 2.07 (s, 3H, NHCOCH3). 13C NMR (126 MHz, CDCl3) δ = 170.9 (NHCOMe), 166.3 и 166.1 (C=O, 2хСОC6H5), 133.9, 133.1, 129.8, 129.63, 129.62, 128.7, 128.6, 128.3 (2хСОC6H5), 80.7 (С-1), 79.1 (C-4), 75.8, 74.1 (C-2, С-3), 62.85 (С-5), 23.5 (NHCOCH3).HRMS (ESI+): m/z calcd for C21H21NO7M+Na+: 422.1216, found 422.1208.
3,5-Di-O-benzoyl-N-propionyl-α-d-xylofuranosylamide(6)from the oxazoline 2:
a2. The N-xylofuranosyl oxazoline derivative 2 (0.08 mg, 0.2 mmol) was kept at 5-8 oC for 5 weeks. The oily residue was chromatographed on a silica gel, using for elution chloroform, chloroform–methanol 11:1 and 5:1 to give (0.072 g, 86%) benzoyl-protected N-propionyl-α-d-xylofuranosyl amide 6.
b2. The oxazoline 2 (0.167 g, 0.42 mmol) after the hydrolysis reaction on silica gel gave 3,5-di-O-benzoyl-N-propionyl-α-d-xylofuranosylamide (6) (0.148 g, 85%) as white solid.M.p. 144-145 oC. (α)D20 +15.1 (c 0.57, CHCl3). IR (KBr): ν 3387, 2924, 1727, 1653, 1525, 1275 сm-1. 1Н NMR (500 MHz, CDCl3) δ = 7.43-8.03 (m, 10H, 2 x СОC6H5), 6.91 (d, 1Н, J = 9.0 Hz, NHCOEt), 6.13 (dd, 1Н, J1,2 = 4.1 Hz, Н-1), 5.57 (dd, 1Н, J3,4 = 3.8, J3,2 = 1.0 Hz, Н-3), 4.77-4.82 (m, 1Н, Н-4), 4.62 (dd, 1Н, Н-5), 4.61 (dd, 1Н, Н-5′), 4.42 (dd, 1Н,Н-2), 2.34 (q, 2H, NHCOCH2CH3). 1.21 (t, 3H, NHCOCH2CH3). 13C NMR (126 MHz, CDCl3) δ = 174.7 (CN), 166.3 and 166.1 (C=O, 2хСОC6H5), 133.9, 133.2, 129.8, 129.7, 129.6, 128.76, 128.7, 128.4 (2хСОC6H5), 80.8 (С-1), 79.1 (C-4), 75.8, 74.1 (C-2, С-3), 62.9 (С-5), 29.8 (NHCOСН2СН3), 9.4 (NHCOСН2СН3). HRMS (ESI+): m/z calcd for C22H23NO7M+Na+: 436.1367, found 436.1366.
5-О-Benzoyl-3-О-p-toluenesulfonyl-N-acetyl-α-D-xylofuranosylamide (7) from the oxazoline 3:
a3.The oxazoline 3 (0.6 g, 1.31 mmol) was coevaporated with chloroform and kept at 5-8 oC for 4 weeks. The residue was treated with methanol under heating and prepared solution was left under cooling. Crystalline product was filtred off and dried on air. 5-О-Benzoyl-3-О-p-toluenesulfonyl-N-acetyl-α-D-xylofuranosylamide (7) (0.375 g, 60%) was prepared as yellow crystals. M.p. 161-163 0С (МеОН).(α)D20 +4.8 (c 0.5, CHCl3). IR (KBr): ν 3394, 1725, 1672, 1516, 1367, 1275, 1182 см-1. 1Н NMR (500 MHz, DMSO-d6) δ: 8.16 (d, 1Н, J 9.5 Hz, NHCOMe), 7.42-7.89 (m, 9H, СОC6H5 and ОSO2C6H4CH3), 6.26 (d, 2-ОН), 5.73 (dd, 1Н, J1,2 3.9 Hz, Н-1), 4.99 (br.s, 1Н, Н-3), 4.47-4.49 (m, 1Н, Н-4), 4.26 (dd, 1Н, Н-5), 4.17 (dd, 1Н, Н-5′), 4.10 (br.s, 1Н,Н-2), 2.33 (s, 3H, ОSO2C6H4CH3), 1.90 (s, 3H, NHCOMe). 13C NMR (126 MHz, DMSO-d6) δ: 170.54 (NHCOMe), 165.68 (C=O, СОC6H5), 145.94, 138.98, 132.82, 130.78, 129.71, 129.65, 129.21, 128.04 (СОC6H5 иОSO2C6H4CH3), 84.63 (С-1), 80.63 (C-4), 74.78, 73.04 (C-2, С-3), 62.54 (С-5), 23.27 (NHCOMe), 21.57 (ОSO2C6H4CH3).HRMS (ESI+): m/z calcd for C21H23NO8S M+Na+: 472.1042, found 472.1035.
b3. The oxazoline 3 (0.554 g, 1.28 mmol)was dissolved in chloroform and placed to the top of a silica gel column which was prepared in chloroform. After 18 h at room temperature a silica gel column was washed chloroform and then further gradual elution with chloroform-methanol 20:1, 15:1 and 9:1 gave (0.490 g, 85%) of 5-О-benzoyl-3-O-tosyl-N-acetyl-α-d-xylofuranosylamide (7).
5-О-Benzoyl-3-О-methanesulfonyl-N-acetyl-α-D-xylofuranosylamide (8) from the oxazoline 4:
b4.The oxazoline 4 (0.354 g, 0.99 mmol) was dissolved in chloroform and placed to the top of a silica gel column which was prepared in chloroform. After 18 h at room temperature a silica gel column was washed chloroform and then further gradual elution with chloroform-methanol 15:1 and 10:1 gave (0.32 g, 86%) of 5-О-benzoyl-3-O-mesyl-N-acetyl-α-d-xylofuranosyamide (8) as oil. (α)D20 +5.6 (c 1.0, CHCl3). IR (solution in CHCl3): ν 3430, 1725, 1687, 1506, 1358, 1178 см-1. 1Н NMR (500 MHz, CDCl3) δ: 7.45-8.05 (m, 5H, СОC6H5), 7.18 (br.d, 1Н, J = 9.0 Hz, NHCOMe), 6.0 (dd, 1Н, J1,2 =3.8 Hz, Н-1), 5.20 (d, 1Н, Н-3), 4.69-4.73 (m, 1Н, Н-4), 4.49-4.55 (m, 3Н, Н-2 and 2H-5), 3.11 (s, 3H, ОSO2CH3), 2.09 (s, 3H, NHCOMe). 13C NMR (126 MHz, CDCl3) δ: 172.1 (NHCOMe), 166.4 (C=O, СОC6H5), 133.4, 129.8, 129.65, 129.5, 128.5 (СОC6H5), 82.9 (С-1), 80.6 (C-4), 75.3, 73.7 (C-2, С-3), 62.2 (С-5), 38.3 (ОSO2CH3).23.5 (NHCOMe). HRMS (ESI+): m/z calcd for C15H19NO8S [M-H2O]+: 355.0726, found 375.0699.
3,5-O-Isopropylidene-N-acetyl-α-d-xylofuranosylamide (10) from the oxazoline 9:
a4. The oxazoline 9 (0.35 g, 1.64 mmol) was coevaporated with chloroform and kept at 5-8 oC for 6 weeks. The oily residue was chromatographed on a silica gel, using for elution mixtures of ethylacetate: petroleum ether, and chloroform-methanol 10:1 to give (0.342 g, 90%) of3,5-O-isopropylidene-N-acetyl-α-d-xylofuranosylamide (10) as oil. (α)D20 +15.7 (c 1.0, CHCl3). IR (solution in CHCl3): ν 3425, 3019, 2932, 1680, 1505, 1377 cm-1. 1Н NMR (500 MHz, CDCl3) δ = 7.26 (br.d, 1H, NH), 5.95 (dd, 1Н, J1,2 = 3.5, J1,NH = 9.7 Hz, Н-1), 4.32 (br.s, 1Н,Н-3), 4.05 (d, 1Н, Н-2), 3.98-4.02 (m, 1Н, Н-4), 3.92 (dd, 1Н, J5,4 = 2.3, J5,5′ = 13.6 Hz, Н-5), 3.84 (d, 1Н, Н-5′), 2.07 (s, 3H, NHCOCH3), 1.45 and 1.38 [2s, 6H, (CH3)2C-]. 13C NMR (126 MHz, CDCl3) δ = 171.7 (NHCOCH3), 97.3 C-(CH3)2, 81.5 (С-1), 75.0 (C-4), 74.9, 71.3 (C-2, С-3), 60.8 (С-5), 23.4 and 19.3 (CH3)2C-, 28.7 (NHCOCH3). HRMS (ESI+):m/z calcd for C10H17NO5M+Na+: 254.1004, found 254.1008.
b5. The oxazoline 9(0.3 g, 1.4 mmol) was dissolved in chloroform and placed to the top of a silica gel column which was prepared in chloroform. After 22 h a silica gel column was washed chloroform and then further gradual elution with chloroform-methanol 30:1, 15:1 and 8:1 gave(0.254 g, 78%) of3,5-O-isopropylidene-N-acetyl-α-d-xylofuranosylamide (10).
N-Acetyl-α-d-xylofuranosylamide(11) from protected N-xylofuranosyl acetamides
c. 3,5-O-isopropylidene-N-acetyl-α-d-xylofuranosylamide (10) (0.250 g, 0.12 mmol) was dissolved in 5 ml 75% aq acetic acid and reaction mixture was stirred for 20 h, then it was coevaporated with toluene (2x10 ml). The residue was chromatographed on a silica gel, using for elution chloroform, chloroform–methanol 10:1, 5:1 to give (0.202 g, 90%) of N-α-d- xylofuranosylacetamide 11.
d1.3,5-Di-О-benzoyl-N-acetyl-α-d-xylofuranosylamide (5) (0.136 g, 0.34 mmol) was dissolved in 14 ml methanol saturated at 0 oC with ammonia, then reaction mixture was stirred for 17 h at room temperature and evaporated to dryness. The residue was chromatographed on a silica gel, using for elution chloroform, chloroform–methanol 15:1, 5:1 to give (0.05 g, 77%) of N-α-d-xylofuranosylacetamide (11). M.p. 148-149 oC. (α)D20 +48.8 (c 0.72, MeOH). IR (KBr): ν 3411, 3368, 1656, 1510, 1301, 1083 сm-1. 1Н NMR (500 MHz, CD3OD) δ = 5,84 (d, 1Н, J1,2 = 3.9 Hz, Н-1), 4.18-4.21 (m, 1Н, Н-4), 4.16 (dd, 1Н, J3,4 = 3.4 Hz, J3,2 = 1.7 Hz, Н-3), 4,02 (dd, 1Н, J2,1 = 1.7 Hz, Н-2), 3.77 (dd, 1Н, J5,4 = 4.9 Hz, J5,5′ = 11.5 Hz, Н-5), 3.73 (dd, 1Н, J5′,4 = 5.2 Hz, Н-5′), 2.03 (s, 3H, NHCOCH3). 13C NMR (126 MHz, CD3OD) δ = 173.8 (NHCOMe), 82.0 (С-1), 81.3 (C-4), 77.8, 77.2 (C-2, С-3), 61.9 (С-5), 22.9 (NHCOMe). HRMS (ESI+): m/z calcd for C7H13NO5M+Na+: 214.0691, found 214.0686.
N-Propionyl-α-d-xylofuranosylamide (12)
d2.3,5-Di-О-benzoyl-N-propionyl-α-d-xylofuranosylamide (6) (0.055 g, 0.13 mmol) was dissolved in 3 ml methanol and 7 ml methanol saturated at 0 oC with ammonia was added to prepared solution, then reaction mixture was stirred for 18 h at room temperature and evaporated to dryness. The residue was chromatographed on a silica gel, using for elution chloroform, chloroform –methanol 20:1, 6:1 to give (0.022 g, 81%) of N-propionyl-α-d-xylofuranosylamide (12) as oil. (α)D20 +44.8 (c 0.53, MeOH). IR (KBr): ν 3400, 3365, 1657, 1505, 1308, 1083 см-1. 1Н NMR (500 MHz, CD3OD) δ: 5.85 (d, 1Н, J1,2 = 3.9 Hz, Н-1), 4.18-4.21 (m, 1Н, Н-4), 4.16 (dd, 1Н, J2,3 = 1.6 Hz, J3,4 = 3.6 Hz, Н-3), 4.02 (dd, 1Н, J2,3 = 1.6 Hz, J2,1 = 3.9 Hz, Н-2), 3.77 (dd, 1Н, J5,4 = 5.0 Hz, J5,5′ = 11.5 Hz, Н-5), 3.73 (dd, 1Н, J5′,4 = 6.2 Hz, Н-5′), 2.31 (q, 2Н, -N=C-СН2СН3), 1.15 (t, 3H, -N=C-СН2СН3). 13C NMR (126 MHz, CD3OD) δ: 175.9 (NHCOEt), 80.5 (С-1), 79.9 (C-4), 76.4, 75.8 (C-2, С-3), 60.4 (С-5), 28.8 (-NHCOСН2СН3), 8.6 (-NHCOСН2СН3).HRMS (ESI+): m/z calcd for C8H15NO5M+Na+: 228.0842, found 228.0843.
Synthesis of N-acyl- α -d-xylofuranosylamides from D-xylose:
N-Acetyl-α-d-xylofuranosylamide (11) from D-xylose
f1. To a stirred solution of D-xylose (0.211 g, 1.4 mmol) in anhydrous acetonitrile (6 ml), KHF2 (0.411 g, 5.24 mmol), boron trifluoride diethyl etherate (1.4 ml, 11.0 mmol) were added at rt. The reaction mixture was stirred at room temperature for 3 h 30 min, and then poured into cooled 25.5 ml aq 1N NaOH and evaporated to dryness, coevaporated with ethanol. The residue was chromatographed on a silica gel, using for elution mixtures of chloroform-methanol 7:1, 6:1 and 4:1 to give 0.027 g of a mixture of 11 and 13 (about 6% yield of 13 according to 1H NMR data) as oil. 1Н NMR (500 MHz, CD3OD) a separate fraction containing a mixture of N-xylosides 11 and 13 (a ratio-2:3.2). 1Н NMR (500 MHz, СD3OD) N-α-D-xylopyranosyl acetamide 13, δ = 5.38 (d, 1Н, J1,2 = 3.1 Hz, Н-1), 3,84 (d, 1Н, J2,1 = 3.1 Hz, Н-2), 3.78-3.81 (m, 1Н, Н-3), 3.58 (dd, 1Н, J5,4 = 5.4 Hz, J5,5′ = 11.8 Hz, Н-5), 3.53 (dd, 1Н, J5′,4 = 3.2 Hz, Н-5′), 3.49-3.51 (m, 1Н,Н-4), 2.0 (s, 3H, NHCOCH3). 13C NMR (126 MHz, СD3OD) δ: 174.2 (NHCOMe), 81.3 (С-1), 77.5, 71.7, 70.3 (C-4, C-2, С-3), 66.4 (С-5), 22.7 (NHCOMe).
1Н NMR (500 MHz, СD3OD) N-α-D-xylofuranosyl acetamide 11, δ = 5,84 (d, 1Н, J1,2 = 3.9 Hz, Н-1), 4.18-4.21 (m, 1Н, Н-4), 4.16 (dd, 1Н, J3,4 = 3.4 Hz, J3,2 = 1.7 Hz, Н-3), 4,02 (dd, 1Н, J2,1 = 1.7 Hz, Н-2), 3.77 (dd, 1Н, J5,4 = 4.9 Hz, J5,5′ = 11.5 Hz, Н-5), 3.73 (dd, 1Н, J5′,4 = 5.2 Hz, Н-5′), 2.03 (s, 3H, NHCOCH3). 13C NMR (126 MHz, CD3OD) δ = 173.8 (NHCOMe), 82.0 (С-1), 81.3 (C-4), 77.8, 77.2 (C-2, С-3), 61.9 (С-5), 22.9 (NHCOMe). HRMS (ESI+): m/z calcd for C7H13NO5M+Na+: 214.0691, found 214.0686.
and 0.099 g (37%) of N-acetyl-α-d-xylofuranosylamide(11) as crystalline product.
N-Propionyl- α -d-xylofuranosylamide ( 12 ) from D-xylose
f2. To a stirred suspension of dried D-xylose (0.218 g, 1.45 mmol) in anhydrous propionitrile (6 ml), KHF2 (424 mg, 5.4 mmol) boron trifluoride diethyl etherate (1.4 ml, 11.0 mmol) were added at rt. The reaction mixture was stirred at room temperature for 4 h, and then poured into cooled 25 ml aq. 1N NaOH and evaporated to dryness. The residue was chromatographed on a silica gel, using for elution mixtures of chloroform-methanol 6:1, 5:1 and 3:1 to give (0.082 g, 28%) of N-propionyl-α-d-xylofuranosylamide(12) as oil.
Benzoylation of 3,5-di-O-benzoyl-N-α-D-xylofuranosyl acetamide 5
g1. To a stirred solution of selectively protected N-xylofuranosyl amide 5 (0.034 g, 0.085 mmol) in anhydrous pyridine (3 ml) benzoyl chloride (0.08 ml, 0.69 mmol) was added at 0 oС and then the reaction mixture was stirred for 48 h at room temperature, diluted with CH2Cl2 and poured into cold 5% aq NaHCO3. The aqueous phase was extracted with CH2Cl2 (3x50 ml), the combined organic extracts were washed cooled 5% aq NaHCO3, water, dried and evaporated. The residue was chromatographed on a silica gel, using for elution mixtures of hexane-ethylacetate 6:1, 5:1, and 2:1 to give (0.022 g,42%) of perbenzoylated N-α-xylofuranosyl amide 14. M.p. 89-92 oC. (α)D20 +78.4 (c 0.7, CHCl3). 1Н NMR (500 MHz, CDCl3) δ = 7.34-8.13 (m, 25H, 4 x СОC6H5), 6.52 (dd, 1Н, J3,2 = 6.0, J3,4 = 7.1 Hz, Н-3), 6.39 (d, 1Н, J1,2 = 7.6 Hz, Н-1), 5.69 (dd, 1Н, J2,1 = 7.6, J2,3 = 6.0 Hz, Н-2), 5.11-5.22 (m, 1Н, Н-4), 4.49 (dd, 1Н, Н-5), 4.60 (dd, 1Н, Н-5′), 2.07 (s, 3H, NHCO(C6H5)CH3. 13C NMR (126 MHz, CDCl3) δ = 173.5, 172.8, 171.4, 166.0 and 165.6 (C=O, CON(C6H5)(CH3, 4хСОC6H5), 133.8, 133.7, 133.5, 133.29, 133.22, 130.2, 129.9, 129.8, 129.5, 129.4, 128.8, 128.5, (5хСОC6H5), 85.4 (С-1), 77.6 (C-4), 77.0, 76.5 (C-2, С-3), 63.4 (С-5), 26.2 (NHCO(C6H5)CH3. HRMS (ESI+): m/z calcd for C35H29NO9M+Na+: 630.1735, found 630.1742.
and (0.018 g, 42%) of tri-O-benzoyl derivative 15. M.p. 54-55 oC. (α)D20 +31.8 (c 0.4, CHCl3). 1Н NMR (500 MHz, CDCl3) δ = 7.42 - 8.1 (m, 15H, 3xСОC6H5),6.41 (dd, 1Н, J1,2= 4.1, JNH,H-1= 9.7 Hz, Н-1), 6.23 (d, 1H, NHCOCH3), 5.87 (dd, 1Н, J3,4 = 4.3, J3,2 = 2.0 Hz, Н-3), 5.67 (dd, 1Н, J2,3 = 2.0, J2,1 = 4.1 Hz, Н-2), 4.85-4.88 (m, 1Н, Н-4), 4.59 (dd, 1Н, Н-5), 4.54 (dd, 1Н, Н-5′), 2.01 (s, 3H, NHCOCH3). 13C NMR (126 MHz, CDCl3) δ = 170.1, 166.2, 164.9, 164.4 (C=O, CONHCH3, 3хСОC6H5), 134.2, 133.9, 133.3, 130.0, 129.9, 129.7, 128.9, 128.7, 128.8, 128.5, 128.4 (3хСОC6H5), 79.8 (С-1), 76.4 (C-4), 75.57, 75.55 (C-2, С-3), 62.6 (С-5), 23.6 (NHCOCH3). HRMS (ESI+): m/z calcd for C28H25NO8M+Na+: 526.1473, found 526.1478.
N-Acetyl-β-d-arabinofuranosylamide (19)from benzoylated N-arabinofuranosyl oxazoline 17:
Preparation of 3,5-di-О-benzoyl-N-acetyl-β-d-arabinofuranosylamide (18)from oxazoline 17
a.The benzoylated N-β-D-arabinofuranosyl oxazoline 17 (0.13 g, 0.95 mmol) gave (0.114 g, 84%) 3,5-di-О-benzoyl-N-acetyl-α-d-arabinofuranosylamide (18) as oil after the hydrolysis reactionon silica gel. (α)D20 +15.8 (c 1.0, CHCl3). IR (KBr): ν 3388, 1722, 1656, 1526, 1271, 1180, 1106 см-1. 1Н NMR (500 MHz, CDCl3) δ: 7.36-8.04 (m, 10H, 2 x СОC6H5), 6.90 (br.d, 1Н, J = 9.1 Hz, NHCOMe), 5.91 (dd, 1Н, J1,2 = 4,0 Hz, Н-1), 5.52 (d, 1Н, J3,2 = 2.8 Hz, Н-3), 4.61 (dd, 1Н, Н-5), 4.58 (dd, 1Н, Н-5′), 4.34-4.38 (m, 1Н, Н-4), 4.31 (br.d, 1Н, Н-2), 2.04 (s, 3H, NHCOCH3). 13C NMR (126 MHz, CDCl3) δ: 170.9 (NHCOMe), 166.3 and 166.3 (C=O, 2хСОC6H5), 133.7, 133.2, 129.8, 129.7, 128.5, 128.6, 128.4 (2хСОC6H5), 81.0 (С-1), 80.5 (C-4), 78.45, 78.42 (C-2, С-3), 64.4 (С-5), 23.4 (NHCOCH3). HRMS (ESI+): m/z calcd for C21H21NO7M+Na+: 422.1216, found 422.1208.
N-Acetyl-β-d-arabinofuranosylamide (19)
b.3,5-Di-О-benzoyl-N-acetyl-α-d-arabinofuranosylamide (18) (0.17 g, 0.42 mmol) was dissolved in 8 ml methanol saturated at 0 oC with ammonia, then reaction mixture was stirred for 18 h at room temperature and evaporated to dryness. The residue was chromatographed on a silica gel, using for elution chloroform, chloroform –methanol 15:1, 6:1 to give (0.028 g, 68%) of N-α-arabinofuranosylacetamide 19. M.p. 148-149 0C. (α)D20 +34.4 (c 0.75, MeOH). IR (KBr): ν 3411, 3368, 1656, 1510, 1301, 1083 см-1. 1Н NMR (500 MHz, CD3OD) δ: 5.73 (d, 1Н, J1,2 = 4.2 Hz, Н-1), 4.0 (t, 1Н, J3,4 = 3.2 Hz, J3,2 = 3.1 Hz, Н-3), 3.92 (dd, 1Н, J2,1 = 4.2 Hz, Н-2), 3.76-3.79 (m, 1Н, Н-4), 3.71 (dd, 1Н, J5,4 = 4.5 Hz, J5,5′ = 11.2 Hz, Н-5), 3.66 (dd, 1Н, J5′,4 4.7 Hz, Н-5′), 2.03 (s, 3H, NHCOCH3).13C NMR (126 MHz, CD3OD) δ:172.3(NHCOMe),83.9 (С-1), 80.7 (C-4), 76.7, 76.6 (C-2, С-3), 62.0 (С-5), 21.5 (NHCOMe). HRMS (ESI+): m/z calcd for C7H13NO5M+Na+: 214.0691, found 214.0686.
Synthesis of N-acetyl-β-d-arabinofuranosylamide 19 and N-acetyl-β-d-arabinopyranosylamide 20 from D-arabinose
c.To a stirred suspension of dried D-arabinose (0.257 g, 1.7 mmol) in anhydrous acetonitrile (8 ml), KHF2 (0.485 g, 6.2 mmol) and boron trifluoride diethyl etherate (1.6 ml, 12.6 mmol) were added at rt. The reaction mixture was stirred at room temperature for 4 h 10 min, and then poured into cooled 28 ml aq. 1N NaOH and evaporated, coevaporated with ethanol to dryness. The residue was dissolved in methanol, chloroform-methanol- 4:1 under mild heating, inorganic salts were filtered off and filtrate was evaporated. The residue was chromatographed on a silica gel, using for elution chloroform, chloroform –methanol 6:1, 5:1 and 4:1, 3:1 to give 0.07 g (21%) of N-acetyl-β-d-arabinofuranosylamide(19) as crystalline product. Spectral data of the latter were identical to those for 19 (CD3OD) prepared from the protected oxazoline 17.
1Н NMR (500 MHz, D2O) δ: 5.59 (d, 1Н, J1,2 = 4.9 Hz, Н-1), 4.1 (t, 1Н, J3,4 = 3.2 Hz, J3,2 = 3.1 Hz, Н-3), 3.98 (dd, 1Н, J2,1 = 4.9 Hz, Н-2), 3.70-3.81 (m, 1Н, Н-4), 3.64 (dd, 1Н, J5,4 = 4.5 Hz, J5,5′ = 11.2 Hz, Н-5), 3.60 (dd, 1Н, J5′,4 4.7 Hz, Н-5′), 1.99 (s, 3H, NHCOCH3). 13C NMR (126 MHz, D2O) δ: 175.0 (NHCOMe), 82.4 (С-1), 80.0(C-4), 75.7, 75.0 (C-2, С-3), 61.4 (С-5), 22.1 (NHCOMe). HRMS (ESI+): m/z calcd for C7H13NO5M+Na+: 214.0691, found 214.0686.
0.058 g (18%) of N-acetyl-β-d-arabinopyranosylamide(20) as white solid.M.p. 167-170 oC. (α)D20 -14.8 (c 0.32, MeOH). 1Н NMR (500 MHz, D2O)δ = 5.23 (d, 1Н, J1,2 = 3.1 Hz, Н-1), 3.82-3.84 (m, 1Н, Н-4), 3.77 (dd, 1Н, J = 3.2 Hz, J3,2 = 6.8 Hz, Н-3), 3.73 (dd, 1Н, J2,1 = 3.2 Hz, Н-2), 3.57 (dd, 1Н, J5,4 = 3.7 Hz, J5,5′ = 12.1 Hz, Н-5), 3.49 (dd, 1Н, J5′,4 = 7.0 Hz, Н-5′), 1.88 (s, 3H, NHCOCH3). 13C NMR (126 MHz, CD3OD) δ = 175.4 (NHCOMe), 75.9 (С-1), 69.2, 68.1, 65.5, (C-4, C-2, С-3), 63.6 (С-5), 21.8 (NHCOMe). HRMS (ESI+): m/z calcd for C7H13NO5M+Na+: 214.0691, found 214.0692.
Preparation of 2-phenyl-β-d-arabinofurano-(1,2-d)-2-oxazoline (24) and 2-phenyl-β-d-arabinopyrano-(1,2-d)-2-oxazoline (25) from D-arabinose
b. To a suspension of dried D-arabinose (0.317 g, 2.11 mmol) in anhydrous benzonitrile (4.5 ml), KHF2 (0.598 g, 7.65 mmol) boron trifluoride diethyl etherate (1.6 ml, 15.6 mmol) were added at rt. The reaction mixture was stirred at room temperature for 5 h 30 min, and poured into cooled 36 ml aq.1N NaOH, prepared solution left at 5-8 0C for 18 h, then coevaporated with ethanol at mild heating to dryness. The residue was dissolved in methanol, chloroform-methanol- 4:1 under mild heating, inorganic salts were filtered off and filtrate was coevaporated with silica gel and placed to the top of a silica gel column which was washed chloroform (60 ml) and then further gradual elution with chloroform-methanol 30:1, 15:1 and 10:1, 5:1 gave 0.338 g of a mixture of products. Additional chromatography on silica gel prepared in chloroform using for elution chloroform and chloroform-petroleum ether-methanol 15:6:0.5 → 15:6:1.5 gave (0.276 g, 56%) of 2-phenyl-β-d-arabinofurano-(1,2-d)-2-oxazoline(24) as oil. Spectral data of the latter were identical to those for 24 prepared from the protected oxazoline 22.
1Н NMR (500 MHz, D2O) δ = 7.42-7.99 (m, 5H, -N=C-C6H5), 6.11 (d, 1Н, J1,2 = 6.2 Hz, Н-1), 5.05 (dd, 1Н, J2,3 = 1.3 Hz, Н-2), 4.33 (br.d, 1Н,Н-3), 3.98-4.0 (m, 1Н, Н-4), 3.48 (dd, 1Н, J5,4 = 6.0, J5,5′ = 11.8 Hz, Н-5), 3.44 (dd, 1Н, J5′,4 = 6.1 Hz, Н-5′). 13C NMR (126 MHz, D2O) δ = 168.7 (CN), 134.0, 130.0 and 128.0 (N-C6H5), 102.0 (С-1), 90.8 (C-4), 87.3, 77.8 (C-2, С-3), 62.9 (С-5). HRMS (ESI+): m/z calcd for C12H13NO4M+H+: 236.0923, found 236.0927.
and (0.050 g, 10 %)2-phenyl-β-d-arabinopyrano-(1,2-d)-2-oxazoline (25) as foam.(α)D20 -21.0 (c 0.5, MeOH). 1Н NMR (500 MHz, D2O) δ =7.37-7.78 (m, 5H, -N=C-C6H5), 5.67 (d, 1Н, J1,2 = 6.8 Hz, Н-1), 4.55 (t, 1Н, J2,3 = 6.7 Hz, Н-2), 3.86-3.88 (m, 1Н, Н-4), 3.76-3.81 (m, 1Н, Н-3 and H-5), 3.61 (dd, 1Н, J5′,4 = 3.2 Hz, J5,5′ = 12.5 Hz, Н-5′).13C NMR (126 MHz, D2O) δ = 168.5 (CN), 133.2, 129.5, 128.7, 128.4 (N-C6H5), 92.5 (С-1), 90.8, 69.2, 66.4 (C-4, C-2, С-3), 66.0 (С-5). LC-MS (ESI+): m/z calcd for C12H13NO4M+H+: 236.1, found 236.1.
2-Phenyl-(3,5-O-1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-β-d-arabinofurano)-(1,2-d)-2-oxazoline(27)
c.To a stirred solution of the oxazoline 24 (0.049 g, 0.21 mmol) in anhydrous pyridine (2.6 ml) 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane (0.132 ml, 4.16 mmol) was added and then the reaction mixture was stirred for 48 h at room temperature. Water was added to prepared solution, the aqueous phase was extracted with CH2Cl2 (3x40 ml), the combined organic extracts were washed with 1N aq. HCl (2x6 ml), cooled 5% aq NaHCO3, water, dried over anh. Na2SO4 and evaporated to dryness. The residue was chromatographed on a silica gel, using for elution a mixture of hexane-ethylacetate 9:1, 8:1, and 5:1 to give (0.097 g,98%) of oxazoline derivative 27 as oil.(α)D20 -31.3 (c 0.8, CHCl3).1Н NMR (500 MHz, CDCl3) δ:= 8.0 (d, 2H, N=C-C6H5), 7.51-7.54 (t, 1H, N=C-C6H5), 7.42 (t, 2H, N=C-C6H5), 6.05 (d, 1Н, J1,2 =6.6 Hz, Н-1), 5.0 (dd, 1Н, J2,3 = 4.1 Hz, Н-2), 4.31 (dd, 1Н, J3,4 = 8.0 Hz, Н-3), 4.05 (dd, 1Н, J5,4 = 3.2, J5,5́ = 12.2 Hz, H-5), 3.91 (dd, 1Н, J5´,4 = 5.9 Hz, Н-5´), 3.23-3.75 (m, 1Н, Н-4), 0.97-1.20 (m, 28Н, 4x (СН3)2CH]. 13C NMR (126 MHz, CDCl3) δ = 166.2 (CN), 132.2, 128.9, 128.4, 127.1 (-N=C-C6H5), 99.6.1 (С-1), 88.9 (C-4), 80.0, 79.9 (C-2, С-3), 62.8 (С-5), 17.5, 17.3, 17.2, 16.89,17.1 [4x(СН3)2CH], 13.3, 13.2, 12.69, 12.66 [4x(СН3)2CH]. HRMS (ESI+): m/z calcd for C24H39N1Si2O5M+H+: 477.2367,found 478.2380.
2-Phenyl-(3,5-di-O-diisopropylsilylhydroxy-β-d-arabinofurano)-(1,2-d)-2-oxazoline(28)
d.To the oxazoline 27 (0.022 g, 0.045 mmol) was added CH2Cl2 (0.5 ml) containing aq.33% HCl (0.027 ml) and then the reaction mixture was stirred for 24 h at room temperature. Water was added to prepared solution, the aqueous phase was extracted with CH2Cl2 (3x40 ml), the combined organic extracts were washed with cooled 5% aq NaHCO3, water, dried over anh. Na2SO4 and evaporated to dryness. The residue was chromatographed on a silica gel, using for elution a mixture of hexane-ethylacetate 9:1, 8:1, and 4:1 to give (0.02 g,88%) of oxazoline derivative 28 as oil.1Н NMR (500 MHz, CDCl3), δ= 7.96 (dd, 2H, N=C-C6H5), 7.50 (t, 1H, N=C-C6H5), 7.43 (t, 2H, N=C-C6H5), 6.17 (d, 1Н, J1,2 =6.3 Hz, Н-1), 4.96 (dd, 1Н, J2,3 = 2.1, J2,1 = 6.3 Hz, Н-2), 4.68 (dd, 1Н, J3,4 = 4.8 Hz, Н-3), 3.93-3.96 (m, 1Н, Н-4), 3.82 (dd, 1Н, J5,4 = 8.0, J5,5́ = 11.5 Hz, H-5), 3.7 (dd, 1Н, J5´,4 = 3.7 Hz, Н-5´), 0.97-1.12 (m, 28Н, 4x (СН3)2CH]. 13C NMR (126 MHz, CDCl3) δ = 166.4 (CN), 132.4, 129.0, 128.4, 126.5 (-N=C-C6H5), 100.4 (С-1), 89.5 (C-4), 84.4, 76.4 (C-2, С-3), 60.8 (С-5), 17.4, 17.2, 17.1, 17.0,16.9 [4x(СН3)2CH], 13.5, 13.4, 13.1, 12.6 [4x(СН3)2CH]. LC-MS (ESI+): m/z calcd forC24H41N1Si2O6M+:478.24, found 478.3.
2-Phenyl-(3,5-di-O-4-chlorobenzoyl)-β-d-arabinofurano)-(1,2-d)-2-oxazoline(29)
e. To a stirred solution of the oxazoline 24 (0.076 g, 0.32 mmol) in anhydrous pyridine (4 ml) 4-chlorobenzoyl chloride (0.17 ml, 1.27 mmol) was added at 0 oС and then the reaction mixture was stirred for 18 h at room temperature, diluted with CH2Cl2 and poured into cold 5% aq NaHCO3. The aqueous phase was extracted with CH2Cl2 (3x50 ml), the combined organic extracts were washed with 1N aq. HCl, cooled 5% aq NaHCO3, water, dried and evaporated. The residue was chromatographed on a silica gel, using for elution a mixture of hexane-ethylacetate 6:1, 5:1, and 3:1 to give (0.147 g,89%) of benzoylated oxazoline derivative 29 as white solid.M.p. 49-52 oC. (α)D20 -124.9 (c 0.67, CHCl3).1Н NMR (500 MHz, CDCl3), δ = 7.99 (d, 4H, 4ClC6H5СО and N=C-C6H5), 7.86 (d, 2H, 4ClC6H5СО), 7.56 (t, 1H, N=C-C6H5), 7.42-7.46(m, 4H, 4ClC6H5СО andN=C-C6H5), 7.22(d, 2H,4-ClСОC6H5),6.4 (d, 1Н, J1,2 = 6.2 Hz, Н-1), 5.67 (br.d, 1Н, J3,4 = 2.3 Hz, Н-3), 5.22 (dd, 1Н, J2,3 = 0.9 Hz, Н-2), 4.56-4.59 (m, 1Н, Н-4). 4.41 (dd, 1Н, J5,4 = 5.5, J5,5́ = 11.7 Hz, H-5), 4.35 (dd, 1Н, J5´,4 = 6.2 Hz, Н-5´). 13C NMR (126 MHz, CDCl3) δ = 166.7 (CN), 165.2 and 164.7 (C=O, 2х4-ClСОC6H5), 140.4, 139.6, 132.7, 131.3, 131.1, 129.1, 129.0, 128.7, 128.6 (2х4-ClСОC6H5, -N=C-C6H5), 102.1 (С-1), 86.3 (C-4), 81.4, 79.5 (C-2, С-3), 63.8 (С-5). HRMS (ESI+): m/z calcd for C26H19NCl2O6 M+H+:512.0663, found 512.0693
3,5-Di-О-4-chlorobenzoyl-N-benzoyl-β-d-arabinofuranosylamide(32)
g. To a stirred solution of benzoylated oxazoline derivative 29 (0.047 mg, 0.09 mmol) in anhydrous acetonitrile (4 ml) 46% aq. HBF4 (0.047 ml, 0.246 mmol) was added at at 0oC. The reaction mixture was stirred under cooling for 30 min and at room temperature for 20 h, and then diluted with CH2Cl2 (5 ml), cold 5% aq NaHCO3 was addedto prepared solution under stirring. The aqueous phase was extracted with CH2Cl2 (3x30 ml). The combined organic extracts were washed with water, dried over anh. Na2SO4, and evaporated to dryness. The residue was chromatographed on a silica gel, using for elution mixtures of hexane-ethylacetate 6:1, 4:1, 1:1 and ethylacetate to give 0.007 g (19%) of cyclic product 31 as a colorless oil. 1Н NMR (500 MHz, CDCl3),δ= 7.22 – 8.02 (4m, 15H, Ph, 4ClPh), 5.82-5.87 (m, 0.7H, NН), 5.69 (s, 1Н,Н-3), 5.51-5.56 (m, 2.3H, H-2 and H-1), 4.82 (dd, 1Н, J5,4 = 3.4, J5,5́ = 11.8 Hz, H-5), 4.72-4.75 (m, 1.4Н, Н-4), 4.64 (dd, 1Н, J5´,4 = 5.2 Hz, Н-5´), 3.2 (br.s, 1H, OH). 13C NMR (126 MHz, CDCl3) δ = 165.5, 165.4 and 165.0 (C=O, 4ClPhCO, CO(NH)-), 140.2, 139.6, 133.7, 131.3, 131.2, 131.14, 131.2, 130.0, 129.9, 128.8, 128.7, 128.5 (4ClC6H5СО, -NH-C-C6H5), 100.9 (С-1), 82.3, 81.5, 70.0 (C-4, C-2 and С-3), 63.9 (С-5). LC-MS (ESI+): m/z calcd for C26H21O7N1Cl2 M+Na+: 552.1, found 553.1.
0.011 g (23%) of the starting oxazoline 29 and 3,5-di-О-4-chlorobenzoyl-N-benzoyl-β-d-arabinofuranosylamide(32)(0.012 g, 32%) as oil. (α)D20 -12.5 (c 0.2, CHCl3).1Н NMR (500 MHz, CDCl3) δ = 7.97 (d, 2H, 4-ClСОC6H5), 7.96(d, 2H, 4-ClСОC6H5), 7.84 (d, 2H, СОC6H5), 7.84 (d, 2H, СОC6H5), 7.54 (t, 1H, СОC6H5), 7.36-7.45 (2m, 6H, 4-ClСОC6H5 and СОC6H5), 6.11 (dd, 1Н, JNH, H-1 = 8.7 Hz, J = 4.4 Hz, Н-1), 5.22 (dd, 1Н, J3,2 = 1,9 Hz, J3,4 = 4,1 Hz, Н-3), 5.52 (d, 1Н, J3,2 = 2.8 Hz, Н-3), 4.64 (d, 2Н, Н-5 and Н-5′), 4.44 (dd, 1Н, J2,1 = 4.4 Hz, Н-2), 4.36-4.41 (m, 1Н, Н-4), 3.4 (br.s., 1H, 2-OH). 13C NMR (126 MHz, CDCl3) δ: 167.6, 165.7 and 165.6 (NHCOBz), 2хСО-4-ClC6H5), 131.28, 131.26, 131.17, 129.1, 129.0, 128.9, 128.6, 127.4 (2хСО4-ClC6H5), 81.5 (С-1), 81.0 (C-4), 78.2, 74.8 (C-2, С-3), 64.4 (С-5), 31.0 (NHCOBz).LS-MS (ESI+): m/z calcd forC26H21O7NCl2M+H+:530.1, found 530.1.
3,5-Di-О-benzoyl-N-acetyl-α-d-ribofuranosylamide(37) from the oxazoline 33:
a. The oxazoline 33 (0.3 g, 0.75 mmol) was coevaporated with chloroform and kept at 5-8 oC for 6 weeks. The oily residue was chromatographed on a silica gel, using for elution mixtures of ethylacetate: petroleum ether, and ethylacetate-methanol 6:1 to give (0.188 g, 62%) of 3,5-di-О-benzoyl-N-acetyl-α-d-ribofuranosylamide (37). M.p. 155-156 0C. (α)D20 +62.2 (c 0.45, CHCl3). 1Н NMR (500 MHz, CDCl3) δ:7.41-8.06(m, 10H, 2 x СОC6H5), 6.84 (d, 1Н, J 8.9 Hz, NHCOMe), 5.97 (dd, 1Н, J1,2 4.4 Hz, Н-1), 5.38 (dd, 1Н, J3,4 4.9 Hz, J3,2 6.5 Hz, Н-3), 4.61-4.65 (m, 2Н, H-2 and Н-4), 4.56 (dd, 1Н, Н-5), 4.51 (dd, 1Н, Н-5′), 2.06 (s, 3H, NHCOCH3). 13C NMR (126 MHz, CDCl3) δ:170.83 (NHCOMe), 166.31 and 166.80 (C=O, 2хСОC6H5), 133.77, 133.24, 129.80, 129.73, 128.58, 128.96, 128.65, 128.44 (2хСОC6H5), 80.32 (С-1), 76.81 (C-4), 74.76, 69.67 (C-3, С-2), 64.28 (С-5), 23.49 (NHCOCH3).HRMS (ESI+): m/z calcd for C21H21NO7M+Na+: 422.1216, found 422.1208.
3,5-Di-О-benzoyl-N-acetyl-α-d-ribofuranosylamide(37) and 2,5-di-О-benzoyl-N-acetyl-α-d-ribofuranosylamide (38)
b. The oxazoline 33 (0.207 g, 0.54 mmol) was dissolved in chloroform and placed to the top of a silica gel column which was prepared in chloroform. After 48 h at room temperature a silica gel column was washed chloroform and then further gradual elution with chloroform-methanol 30:1, 15:1 and 8:1 gave 0.16 g of a mixture of protected N-ribofuranosides, The prepared mixture of isomeric N-ribosides was chromatographed on silica gel using using for elution a mixture of hexane-ethylacetate 1:1, 1.5:1 and 1:2 to give (0.032 g,15%)of2,5-di-О-benzoyl-N-acetyl-α-d-ribofuranosylamide(38).M.p. 160-1610C. (α)D20 +18.0 (c 0.13, CHCl3). 1Н NMR (500 MHz, CDCl3) δ: 7.49-8.1 (m, 10H, 2 x СОC6H5), 6.79 (d, 1Н, J = 9.5 Hz, NHCOMe), 6.18 (dd, 1Н, J1,2 = 5.4 Hz, Н-1), 5.46 (t, 1Н, J2,3 = 5.2 Hz, J2,1 = 5.4 Hz, Н-2), 4.66 (m, 1Н, Н-4), 4.54 (dd, 1Н, J5,4 = 4.9 Hz, J5,5′ = 12.9 Hz, Н-5), 4.47-4.52 (m, 2Н, Н-5′ and H-3), 2.06 (s, 3H, NHCOCH3). 13C NMR (126 MHz, CDCl3) δ: 170.5 (NHCOMe), 166.5 and 165.6 (C=O, 2хСОC6H5), 133.9, 133.4, 129.8, 129.7, 129.5, 128.9, 128.7, 128.6 (2хСОC6H5), 81.3 (С-1), 79.0 (C-4), 72.5, 71.6 (C-3, С-2), 64.4 (С-5), 23.6 (NHCOCH3). HRMS (ESI+): m/z calcd for C21H21NO7M+Na+: 422.1216, found 422.1211.
(0.097 g, 45%)of 3,5-di-О-benzoyl-N-acetyl-α-d-ribofuranosylamide(37).
M.p. 155-156 0C. (α)D20 +62.2 (c 0.45, CHCl3). 1Н NMR (500 MHz, CDCl3) δ: 7.41-8.06 (m, 10H, 2 x СОC6H5), 6.84 (d, 1Н, J = 8.9 Hz, NHCOMe), 5.97 (dd, 1Н, J1,2 = 4.4 Hz, Н-1), 5.38 (dd, 1Н, J3,4 = 4.9 Hz, J3,2 = 6.5 Hz, Н-3), 4.61-4.65 (m, 2Н, H-2 and Н-4), 4.56 (dd, 1Н, Н-5), 4.51 (dd, 1Н, Н-5′), 2.06 (s, 3H, NHCOCH3). 13C NMR (126 MHz, CDCl3) δ: 170.8 (NHCOMe), 166.3 and 166.8 (C=O, 2хСОC6H5), 133.7, 133.2, 129.8, 129.7, 128.6, 128.96, 128.6, 128.4 (2хСОC6H5), 80.3 (С-1), 76.8 (C-4), 74.8, 69.7 (C-3, С-2), 64.3 (С-5), 23.5 (NHCOCH3). HRMS (ESI+): m/z calcd for C21H21NO7M+Na+: 422.1216, found 422.1208.
N-Acetyl-α-d-ribofuranosylamide (39)
c. 3,5-Di-О-benzoyl-N-acetyl-α-d-ribofuranosylamide (37) (0.170 g, 0.43 mmol) was dissolved in 15 ml methanol saturated at 0oC with ammonia, then reaction mixture was stirred for 10 h at room temperature and evaporated to dryness. The residue was chromatographed on a silica gel, using for elution chloroform, chloroform –methanol 15:1, 5:1 and 2:1 to give (0.061 g, 72%) of N-α-ribofuranosylacetamide 39 as oil. (α)D20 +61.4 (c 0.33, MeOH). 1Н NMR (500 MHz, CD3OD)δ: 5.68 (d, 1Н, J1,2 4.4 Hz, Н-1), 4.09-4.13 (m, 2Н, Н-2 and H-3), 3.91-3.94 (m, 1Н, Н-4), 3.71 (dd, 1Н, J5,4 3.1 Hz, J5,5′ 12.1 Hz, Н-5), 3.57 (dd, 1Н, J5′,4 4.2 Hz, Н-5′), 2.04 (s, 3H, NHCOCH3). 13C NMR (126 MHz, CD3OD) δ: 173.86 (NHCOMe), 84.18 (С-1), 81.64 (C-4), 72.54, 71.92 (C-2, С-3), 62.96 (С-5), 22.95 (NHCOMe). HRMS (ESI+):m/z calcd for C7H13NO5M+Na+: 214.0691, found 214.0684.
d. To a stirred suspension of dried D-ribose (0.210 g, 1.39 mmol) in anhydrous acetonitrile (4.5 ml), KHF2 (0.328 g, 4.2 mmol) and boron trifluoride diethyl etherate (1.15 ml, 9.0 mmol) were added 00C. The reaction mixture was stirred under cooling for 30 min and then for 3 h at room temperature, and then poured into cooled 25 ml aq. 1N NaOH and evaporated, coevaporated with ethanol to dryness. The residue was dissolved in methanol, chloroform-methanol- 4:1 under mild heating, inorganic salts were filtered off and filtrate was evaporated. The residue was chromatographed on a silica gel, using for elution chloroform, chloroform –methanol 6:1, 5:1 and 3:1, 1:1 to give 0.024 g (9%) of N-acetyl-α-d-ribofuranosylamide(39) as oil. Spectral data of the latter were identical to those for 39 prepared from the protected oxazoline 37.
Synthesis of 3,5-di-О-benzoyl-N-acetyl-α-and β-d-ribofuranosylamides 42αand 43β from peracylated d-ribose 41
e. To a stirred solution of 1-O-acetyl-2,3,5-tri-O-benzoyl-β-d-ribofuranose (41) (0.250 g, 0.495 mmol) in anhydrous acetonitrile (10 ml) KHF2 (0.116 g, 1.48 mmol) and boron trifluoride diethyl etherate (0.36 ml, 2.84 mmol) were added at 00C. Then, the reaction mixture was stirred at room temperature for 2 h, and then poured into cooled 6.5 ml 1N aq NaOH. The aqueous phase was extracted with CH2Cl2 (3x40 ml). The combined organic extracts were washed with water, dried over anh. Na2SO4, and evaporated to dryness. The residue was chromatographed on a silica gel, using for elution mixtures of hexane-ethylacetate 6:1, 4:1 and 1:1 to give 2,3,5-tri-О-benzoyl-N-acetyl-β-d-ribofuranosylamide(43β) (0. 025 g, 10%). (α)D20 -4.1 (c 0.6, CHCl3). 1Н NMR (500 MHz, CDCl3) δ: 7.41-8.15 (m, 15H, 3 x СОC6H5), 6.47 (d, 1Н, J 8.9 Hz, NHCOMe), 6.07 (dd, 1Н, J1,2 6.5 Hz, Н-1), 5.84 (dd, 1Н, J3,2 5.1 Hz, J3,4 3.6 Hz, Н-3), 5.62 (t, 1Н, Н-3), 4.75 (dd, 1Н, Н-5), 4.62-4.65 (m, 1Н, Н-4), 4.61 (dd, 1Н, Н-5′), 2.03 (s, 3H, NHCOCH3). 13C NMR (126 MHz, CDCl3) δ: 170.80 (NHCOMe), 166.23, 165.64 and 165.56 (C=O, 3хСОC6H5), 133.71, 133.66, 133.45, 129.92, 129.86, 129.76, 128.67, 128.55, 128.52 (3хСОC6H5), 82.16 (С-1), 79.28 (C-4), 76.81, 73.88 (C-3, С-2), 64.20 (С-5), 23.45 (NHCOCH3).HRMS (ESI+): m/z calcd for C28H25NO8M+Na+:526.1472, found 526.1423.
and 2,3,5-tri-О-benzoyl-N-acetyl-α-d-ribofuranosylamide (42α) (0.025 g, 10%). (α)D20 +23.8 (c 0.76, CHCl3). 1Н NMR (500 MHz, CDCl3) δ: 7.40-8.12 (m, 15H, 3 x СОC6H5), 6.45 (d, 1Н, J 9.6 Hz, NHCOMe), 6.38 (dd, 1Н, J1,2 4.5 Hz, Н-1), 5. 85-5.88 (m, 2Н, H-2 and Н-3), 4.72-4.74 (m, 1Н, Н-4), 4.67 (dd, 1Н, Н-5), 4.60 (dd, 1Н, Н-5′), 2.08 (s, 3H, NHCOCH3). 13C NMR (126 MHz, CDCl3) δ:169.93 (NHCOMe), 166.22, 165.08 and 164.80 (C=O, 3хСОC6H5), 133.88, 133.77, 133.34, 129.79, 129.57, 128.71, 128.67, 128.56 (3хСОC6H5), 79.08 (С-1), 78.62 (C-4), 72.91, 71.02 (C-3, С-2), 64.25 (С-5), 23.64 (NHCOCH3).HRMS (ESI+):m/z calcd for C28H25NO8M+Na+:526.1472, found 526.1434.
3,5,6-Tri-O-benzoyl-N-acetyl-α-d-glucofuranosylamide(45) from the oxazoline44:
a1. The oxazoline 44 (0.3g, 0.75 mmol) 24 was coevaporated with chloroform and kept at 5-8 oC for 7 weeks. The oily residue was chromatographed on a silica gel, using for elution mixtures of ethylacetate-petroleum ether, and chloroform-methanol 9:1 to give 0.202 g (66%) of benzoylated N-acetyl-α-d-glucofuranosylamide (45) as oil. (α)D20 -57.3 (c 1.0, CHCl3). IR (KBr): ν 3358, 2927, 1727, 1656, 1519, 1281, 1267, 1109 см-1. 1Н NMR (500 MHz, CDCl3) δ = 7.31-7.99 (m, 15H, 3 x СОC6H5), 6.85 (br.d, 1Н, J = 8.8 Hz, NHCOMe), 6.11 (dd, 1Н, J1,2 = 3.8 Hz, Н-1), 5.74-5.78 (m, 1Н, Н-5),5.56 (d, 1Н, J3,4 = 3.3 Hz, Н-3),4.92 (dd, 1Н, J6,5 = 2.5, J6,6′ = 12.3 Hz, H-6), 4.84 (dd, 1Н, Н-4), 4.64 (dd, 1Н, J6,5 = 5.5 Hz, H-6´),4.32 (d, 1Н, J2,1 = 3.7 Hz, Н-2). 13C NMR (126 MHz, CDCl3) δ = 170.9 (CONHCH3), 166.2, 166.0 and 165.1(C=O, 3хСОC6H5), 133.8, 133.2, 132.06, 129.9, 129.7, 129.6, 128.6, 128.4, 128.3(3хСОC6H5 ),81.1 (С-1), 78.4, 76.0, 74.1, 68.3 (C-5, C-4, C-2, С-3), 64.1 (С-6), 29.7(NHCOCH3). HRMS (ESI+): m/z calcd for C29H27NO9 M+Na+: 556.1578, found 556.1554.
a2. The oxazoline 44 (250 mg) was kept at 5-8 oC for a week. The oily residue was chromatographed on a silica gel, using for elution mixtures of ethylacetate: petroleum ether 3:1, 1:1 and ethylacetate-methanol 9:1 to give (0.133 g, 53%) of the oxazoline 44 and 0.085 g (33%) of 3,5,6-tri-O-benzoyl-N-acetyl-α-d-glucofuranosylamide (45) as oil.
b.The oxazoline 44 (0.100 g, 0.25 mmol) was dissolved in chloroform and placed to the top of silica gel column which was prepared in chloroform. After 24 h a silica gel column was washed chloroform and further chromatographygave 0.08 g (78%) of benzoylated N-acetyl-α-d-glucofuranosylamide (45) as oil, using for elution mixtures of chloroform-methanol 20:1, 15:1 and 10:1.
N-Acetyl-α-d-glucofuranosylamide (46):
c. 3,5,6-Tri-О-benzoyl-N-acetyl-α-d-glucofuranosylamide (45) (0.17 g, 0.32 mmol) was dissolved in 7 ml methanol and 11 ml methanol saturated at 0oC with ammonia was added to prepared solution, then reaction mixture was stirred for 18 h at room temperature and evaporated to dryness. The residue was chromatographed on a silica gel, using for elution chloroform, chloroform–methanol 15:1, 6:1 and 2:1 to give (0.046 g, 65%) of N-acetyl-α-d-glucofuranosylamide (46). m.p. 189-190 oC. (α)D20 +91.4 (c 0.65, MeOH). 1Н NMR (500 MHz, CD3OD) δ = 5.85 (d, 1Н, J1,2 = 3.6 Hz, Н-1), 4.24 (dd, 1Н, Н-3), 4.05 (dd, 1Н, Н-4), 4.01 (dd, 1Н, J2,1 = 3.6, J2,3 = 1.1 Hz, Н-2), 3.86-3.90 (m, 1Н, Н-5), 3.78 (dd, 1Н, J6,5 = 3.1, J6,6′ = 11.5 Hz, Н-6), 3.59 (dd, 1Н, J6´,5 = 6.2, Н-6′), 2.03 (s, 3H, NHCOCH3). 13C NMR (126 MHz, CD3OD) δ = 172.3 (NHCOMe), 81.0 (С-1), 79.2, 76.3, 75.5, 69.6 (C-4, C-2, C-3, C-5), 64.0 (С-6), 21.5 (NHCOCH3). HRMS (ESI+):m/z calcd for C8H15NO6M+Na+: 244.0797, found 244.0794.
d. To solution 3,5,6-tri-О-benzoyl-N-acetyl-α-d-glucofuranosylamide (45) (0.16 g, 0.30 mmol) in 2 ml anhydrous methanol and 0.36 ml 1 M methanolic NaOMe solution was added and then the reaction mixture was kept at rt for 14 h. Amberlyt 15 (H+ form) was added to remove sodium ions, the resin was filtered off, and washed with methanol, solvent was removed under diminished pressure. The residue was chromatographed on a silica gel, using conditons described above, to give (0.048 g, 72%) of N-acetyl-α-d-glucofuranosylamide (46).
N-Acetyl-α-d-glucofuranosylamide (46) and N-acetyl-α-d-glucopyranosylamide (51) from D-glucose:
To a stirred suspension of dried D-glucose(0.25 g, 1.38 mmol) in anhydrous acetonitrile (7.5 ml), KHF2 (0.445 g, 5.7 mmol) boron trifluoride diethyl etherate (1.4 ml, 11.0 mmol) were added at rt. The reaction mixture was stirred at room tempera ture for 4 h, and then poured into cooled 25 ml 1N aq NaOH and evaporated to dryness, coevaporated with ethanol. The residue was chromatographed on a silica gel, using for elution mixtures of chloroform-methanol 4:1 3:1 and 1:1 to give 0.115 g (36%) ofN-acetyl-α-d-glucopyranosylamide(51)as oil.1Н NMR (500 MHz, D2O) δ = 5.38 (d, 1Н, J1,2 = 5.6 Hz, Н-1), 3.6 (dd, 1Н, Н-2), 3.48-3.59 (m, 3H, H-6 and H-6’, H-3), 3.32 (ddd, 1Н, J = 2.3, J = 4.9, J = 10.2 Hz, Н-5), 3.24 (dd, 1H, H-4), 1.9 (s, 3H, NHCOCH3). 13C NMR (126 MHz, CD3OD) δ = 175.9 (NHCOMe), 76.4 (С-1), 72.9, 72.5, 69.23, 69.21, 60.8 (C-4, C-2, C-3, C-5), 60.3 (С-6), 21.9 (NHCOMe). (α)D20 +105.4 (c 0.35, MeOH). HRMS (ESI+):m/z calcd for C8H15NO6M+Na+: 244.0797, found 244.0793.
The further elution with a mixture of chloroform: methanol: water - 20:5:1 gave 0.040 g (13%) ofN-acetyl-α-d-glucofuranosylamide (46). Spectral data of the latter were identical to those for 46 (CD3OD) prepared from the protected oxazoline 44.
3,5,6-Tri-O-benzoyl-N-acetyl-α-d-allofuranosylamide(48) from benzoyl-protected N-α-d-allo-furanosyloxazoline47:
2-Methyl-(3,5,6-tri-О-benzoyl-α-d-allofurano)-(1,2-d)-2-oxazoline (47):
To a stirred solution of intermediate 2,3,5-tri-O-benzoylated allofuranose-1,2-acetonide (0.1 g, 0.34 mmol) in anhydrous benzonitrile (3.2 ml) KHF2 (0.050 g, 1.66 mmol) and boron trifluoride diethyl etherate (0.2 ml, 2.83 mmol) were added successively. The reaction mixture was stirred at room temperature for 18 h, and then poured into cooled 3.47 ml 1N aq NaOH. The aqueous phase was extracted with CHCl3 (3x30 ml). The combined organic extracts were washed with water, dried over anh. Na2SO4, and evaporated to give the oxazoline 47 (0.09 g, 93%) as a colorless oil. (α)D20 +68.1 (c 1.0, CHCl3).1Н NMR (500 MHz, CDCl3) δ = 7.31-8.01 (m, 20H, 2 x СОC6H5, -N=C-C6H5), 6.14 (d, 1Н, J1,2 = 4.5 Hz, Н-1), 5.85-5.89 (m, 1Н, Н-5), 5.34 (d, 1Н, J3,2 = 5.8 Hz, J3,4 = 6.0 Hz, Н-3), 5.25 (d, 1Н, J2,1 = 4.5 Hz, Н-2), 4.84 (dd, 1Н, J6,5 = 3.5, J6,6′ = 12.1 Hz, H-6), 4.67 (dd, 1Н, J6,5 = 7.0 Hz, H-6´), 4.20 (dd, 1Н, Н-4). 13C NMR (126 MHz, CDCl3) δ = 166.8 (CN), 166.1, 165.6 and 165.4 (C=O, 3хСОC6H5), 133.4, 133.3, 133.2, 132.7, 129.8, 129.7, 128.4, 128.37 (3хСОC6H5, -N=C-C6H5), 100.6 (С-1), 78.6, 74.9, 74.7, 71.1 (C-5, C-4, C-2, С-3), 63.3 (С-6). HRMS (ESI+):m/z calcd for C29H25NO8M+Na+: 538.1478, found 538.1455.
3,5,6-Tri-O-benzoyl-N-acetyl-α-d-allofuranosylamide(48) by hydrolysis of the oxazoline47
e. The oxazoline 47 (0.07 g, 0.17 mmol) was dissolved in chloroform and placed to the top of silica gel column which was prepared in chloroform. After 18 h a silica gel column was washed chloroform and further chromatographygave 0.051 g (70%) of benzoylated N-acetyl-α-d-allofuranosylamide (48) as oil using for elution mixtures of chloroform-methanol 20:1, 15:1 and 10:1. (α)D20 +38.8 (c 0.33 CHCl3). 1Н NMR (500 MHz, CDCl3) δ = 7.33-7.95 (m, 15H, 3 x СОC6H5), 6.68 (br.d, 1Н, J = 9.0 Hz, NHCOMe), 6.11 (dd, 1Н, J1,2 = 3.8 Hz, Н-1), 5.62-5.68 (m, 1Н, Н-5),5.53 (dd, 1Н, J3,4 = 4.9 Hz, J3,2 = 6.5 Hz, Н-3),4.92 (dd, 1Н, J6,5 = 3.6, J6,6′ = 12.2 Hz, H-6), 4.50-4.64 (m, 3Н, Н-4, H-6´, Н-2). 13C NMR (126 MHz, CDCl3) δ = 170.6 (CONHCH3), 166.1 and 165.6(C=O, 3хСОC6H5), 133.7, 133.3, 133.2, 129.9, 129.7, 128.6, 128.5, 128.4, 128.3(3хСОC6H5 ),80.2 (С-1), 75.2, 71.8, 69.8 (C-5, C-4, C-2, С-3), 64. 63.2 (С-6), 23.6(NHCOCH3). HRMS (ESI+): m/z calcd for C29H27NO9 M+Na+: 556.1578, found 556.1559.
f. The oxazoline 47 (0.045 g) was kept at 5-8 oC for six weeks. The oily residue was chromatographed on a silica gel, using for elution mixtures of ethylacetate-petroleum ether 3:1, chloroform-methanol 15:1 and 9:1 to give (0.039 g, 85%) 3,5,6-tri-O-benzoyl-N-acetyl-α-d-allofuranosylamide (48) as oil.
6-О-benzoyl-3,4-O-isopropylidene-α-d-galactopyranosylamine (55) from the oxazoline53:
2-Methyl-(6-О-benzoyl-3,4-O-isopropylidene-α-d-galactopyrano)-(1,2-d)-2-oxazoline (53)
a. To a stirred solution of 6-O-benzoyl d-galactopyranose diacetonide 52 (0.44 g, 1.23 mmol) in anhydrous acetonitrile (10 ml) KHF2 (0.226 g, 2.35 mmol) and boron trifluoride diethyl etherate (1.1 ml, 8.75 mmol) were added successively. The reaction mixture was stirred at room temperature for 18 h, and then poured into cooled 21.5 ml 1N aq NaOH. The aqueous phase was extracted with CHCl3 (3x50 ml). The combined organic extracts were washed with water, dried over anh. Na2SO4, and evaporated to give the oxazoline 53 (0.41 g, 93%) as a colorless oil. (α)D20 - 66.2 (c 1.0, CHCl3). IR (in CHCl3): ν 2994, 2931, 1722, 1665, 1373, 1273, 1240 cm-1. 1Н NMR (500 MHz, CDCl3) δ = 7.46-8.1 (m, 5H, СОC6H5), 5.83 (d, 1Н, J1,2 = 7.6 Hz, Н-1), 4.76 (dd, 1Н, J2,1 = 7.7, J2,3 = 2.1 Hz, Н-2), 4.52-4.60 (m, 2Н, H-6 and Н-3), 4.45 (dd, 1Н, J6,5 = 7.2, J6,6́ = 11.4 Hz H-6´), 4.34 (dd, 1Н, Н-4), 3.57-3.61 (m, 1Н, Н-5), 2.13 (s, 3H, NCH3), 1.55 and 1.41 (2s, 3H, (CH3)2C-). 13C NMR (126 MHz, CDCl3) δ = 169.0 (CN), 166.4 (C=O, СОC6H5), 133.0, 130.0, 129.8, 128.4 (СОC6H5), 110.0 [C-CH3)2], 91.5 (С-1), 73.0, 70.7, 70.2, 65.8 (C-5, C-4, C-2, С-3), 63.5 (С-6), 26.7 and 24.7 [(CH3)2C-], 13.9 (NMe). HRMS (ESI+):m/z calcd for C18H21NO6M+H+: 348.1442, found 348.1443, and C18H21NO6M+Na+: 370.1261, found 370.1263.
Preparation of 6-O-benzoyl-N-acetyl-3,4-O-isopropylidene-α-d-galactopyranosylamide (55) by hydrolysis of the oxazoline 53
b.The oxazoline 53 (0.32 g, 0.89 mmol) was dissolved in chloroform and placed to the top of silica gel column which was prepared in chloroform. After 24 h a silica gel column was washed chloroform and then further gradual elution with chloroform-methanol 30:1, 15:1 and 8:1chloroform-methanol gave (0.275 g, 82%) ofprotected N-α-d-galactopyranosylamide (55) as oil. (α)D20 +70.1 (c 0.67, CHCl3). IR (solution in CHCl3): ν 2953, 2928, 1721, 1686, 1492, 1378, 1263 cm-1. 1Н NMR (500 MHz, CDCl3) δ = 7.46-8.1 (m, 5H, СОC6H5), 7.17 (br.d, 1H, NH), 5.67 (dd, 1Н, J1,2 = 3.5, J1,NH = 9.7 Hz, Н-1), 4.48-4.52 (m, 5Н, Н-3, H-4, H-5, H-6, H-6’), 4.09 (t, 1Н, Н-2), 2.04 (s, 3H, NHCOCH3), 1.54 and 1.37 [2s, 3H, (CH3)2C-]. 13C NMR (126 MHz, CDCl3) δ = 171.0 (NHCOCH3), 166.4 (C=O, СОC6H5), 133.1, 129.8, 129.6, 128.3 (СОC6H5), 110.3 [C-CH3)2], 74.2 (С-1), 73.5, 72.1, 68.7, 66.8 (C-4, C-2, С-3, C-5), 64.2 (С-6), 26.4 and 24.6 [(CH3)2C-], 23.4 (NHCOCH3). HRMS (ESI+):m/z calcd for C18H23NO7M+Na+: 388.1372, found 318.1368.
N-Acetyl-α-d-galactopyranosylamide (56) from protected N-acetyl-3,4-O-isopropylidene-α-d-galactopyranosylamide (55)
c.The N-acetyl d-galactopyranosylamide deivative 55 (0.25 mg, 0.67 mmol) was dissolved in 4 ml 80% aq acetic acid and reaction mixture was stirred at 50-55 oC for 18 h, then coevaporated with toluene (2x10 ml). Then residue was dissolved in 4 ml methanol and 12 ml methanol saturated at 0 oC with ammonia was added to prepared solution, then reaction mixture was stirred for 18 h at room temperature and evaporated to dryness. The residue was chromatographed on a silica gel, using for elution chloroform, chloroform–methanol 6:1, 3:1 and 2:1, then chloroform–methanol-water 20:5:1 to give (0.085 g, 58%) of N-acetyl-α-d-galactopyranosylamide (56). M.p. 168-170 0C (crystallization under storing). (α)D20 +86.56 (c 1, MeOH). 1Н NMR (500 MHz, D2O) δ = 5.52 (d, 1Н, J1,2 = 5.7 Hz, Н-1), 3.96 (dd, 1Н, Н-2), 3.89 (br.d, 1H, H4), 3.75 (dd, 1Н, Н-3), 3.69 (m, 1Н, Н-5), 3.61 (dd, 2Н, H-6 and H-6́ ), 2.09 (s, 3H, NHCOCH3). 13C NMR (126 MHz D2O) δ = 175.9 (NHCOMe), 76.7 (С-1), 71.8, 69.3, 68.8, 66.1 (C-4, C-2, C-3, C-5), 61.0 (С-6), 21.9 (NHCOMe). HRMS (ESI+):m/z calcd for C8H15NO6M+Na+: 244.0797, found 244.0793.
Preparation of N-Acetyl-α-d-galactopyranosylamide (56) and N-acetyl-α-d-galactofurano-sylamide (57) from D-galactose
d.To a stirred suspension of dried D-galactose(0.27 g, 1.49 mmol) in anhydrous acetonitrile (8 ml), KHF2 (0.47 g, 6.0 mmol) boron trifluoride diethyl etherate (1.5 ml, 11.8 mmol) were added at rt. The reaction mixture was stirred at room temperature for 3 h 30 min, and then poured into cooled 27 ml 1N q NaOH and evaporated to dryness, coevaporated with ethanol. The residue was chromatographed on a silica gel, using for elution chloroform, chloroform-petroleum ether: methanol 4:2:1 3:1, chloroform–methanol 1:1, and methanol to give 0.05 mg (18%) of N-acetyl-α-d-galactofuranosylamide(57) as oil. (α)D20 +24.8 (c 0.56, MeOH).1Н NMR (500 MHz, D2O)δ=5.46 (d, 1Н,J1,2=4.5Hz, Н-1), 3.96-4.0 (m, 2H, H-4, H-5), 3.63 (dt, 1H, J = 4.4, J = 3.3, J = 7.0, Hz, H-3), 3.58 (dd, 1Н, J2,1 = 4.4, J2,3 = 4.1Hz, Н-2), 3.49 (dd, 1H, J6,5 = 4.6 Hz, J6,6´ = 11.7 Hz, H-6), 3.42 (dd, 1H, J6,5´ = 7.3 Hz, H-6´), 1.9 (s, 3H, NHCOCH3). 13C NMR (126 MHz, D2O) δ = 174.9 (NHCOMe), 81.4 (С-1), 79.7, 75.7, 74.8, 70.8 (C-4, C-2, C-3, C-5), 62.4 (С-6), 21.9 (NHCOMe). HRMS (ESI+):m/z calcd for C8H15NO6M+Na+: 244.0797, found 244.0792.
and 0.048 g(15%) ofN-acetyl-α-d-galactopyranosylamide as oil. Spectral data of the latter were identical to those for 56 (1H and 13C NMR in D2O) prepared from the protected oxazoline 53.
Preparation of 6-О-benzoyl-3,4-O-isopropylidene-β-d-galactopyranosylamine(59β) fromthe oxazoline 53
e.The oxazoline 53 (0.062 g) was stored at 5-8 0C for 3 months andβ-d-galactopyranosylamine derivative 59β was prepared as white solid (according to 1H, 13C NMR and 13C/ DEPT NMR data of a mixture in CDCl3, 85% yield of amine 59β was determined from 1H NMR). 1Н NMR (500 MHz, CDCl3) δ = 7.43-8.07 (m, 10H, СОC6H5), 4.87 (dd, 1Н, J2,1 = 8.7, J2,3 = 7.7 Hz, Н-2), 4.61 (dd, 1H, J6a,5 = 4.7, J6a,6́b= 12.3 Hz, H-6a), 4.53 (dd, 1H, J6b,5 = 7.3 Hz, H-6b), 4.26 (dd, 1Н, J4,5 = 2.1, J4,3 = 5.4 Hz, Н-4), 4.21 (dd, 1Н, Н-3), 4.10-4.14 (m, 1Н, Н-5), 4.03 (d, 1H, J = 8.7 Hz, H-1), 2.12 (s, 3H, CH3CO), 1.96 (br.s, 2H, NH2), 1.55 and 1.35 [2s, 3H, (CH3)2C-]. 13C NMR (126 MHz, CDCl3) δ = 170.8 (COCH3), 166.5 (C=O, СОC6H5), 133.2, 129.9, 129.8, 128.4 (СОC6H5), 110.6 [C-CH3)2], 84.2 (С-1), 79.8, 74.0, 73.8, 71.5 (C-2, C-4, С-3, C-5), 64.1 (С-6), 27.8 and 26.3 [(CH3)2C-], 21.2 (COCH3). HRMS (ESI+):m/z calcd for C18H23NO7M+H+: 366.1548, found 366.1539, M+Na+: 388.1372, found 388.1368. LS-MS (ESI+):m/z calcd for C18H23NO7 [M-NH2]+: 349.12, found 349.1, M+Na+: 388.13, found 388.1.
The structure of β-d-galactopyranosylamine derivative 59β was confirmed by 1H and 13C NMR spectral data, 1H-1H COSY and 2D NOESY spectrum (Figure 2). An evident cross-peak between H-1 and H-5 protons observed in the NOESY spectra (Figure 2) provides support of the β-anomeric configuration of 59β. There are cross-peaks between the protons H-1 and H-3, and the protons H-4 and H-5. Besides, a weak NOE effect was observed for H-1 and H-2.
f. The oxazoline 53 (0.1 g, 0.29 mmol) was dissolved in CH2Cl2 (4.0 ml) and then 0.015 ml 33% aq. HCl was added to prepared solution. The reaction mixture was stirred at rt for 48 h, diluted CH2Cl2 and then was washed with cooled 5% aq NaHCO3. The aqueous phase was extracted with CH2Cl2 (2x10 ml). The combined organic extracts were washed with water, dried over anh. Na2SO4, and evaporated to dryness to give (0.095 g) of oil product containing the starting oxazoline and galactopyranosylamine derivative. The formation of N-β-galactopyranosylamine derivative 59β (34%) was determined from 1H NMR spectra of the reaction mixture taken in CDCl3.
Figure 2.1D NOE correlations for β-d-galactopyranosylamine derivative 59β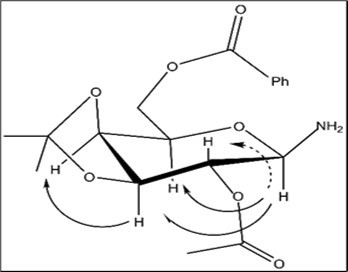
6-O-Benzoyl-N-acetyl-3,4-O-isopropylidene-2-O-acetyl-β-d-galactopyranosylamide(60)
To a stirred solution of protected β-d-galactopyranosylamine59β (0.018 g, 0.049 mmol) and Et3N (0.01 ml, 0.07 mmol) in anhydrous CH2Cl2 (2.0 ml), acetyl chloride (0.02 ml, 0.28 mmol) was added at 00C. The reaction mixture solution was stirred at room temperature for 18 h, diluted CH2Cl2 and then was washed cooled with 5% aq NaHCO3. The aqueous phase was extracted with CH2Cl2 (2x10 ml). The combined organic extracts were washed with water, dried over anh. Na2SO4, and evaporated to dryness. The residue was chromatographed on a silica gel, using for elution chloroform, chloroform–ehylacetate 1:1 to give (0.014 g, 75%) of N-β-d-galactopyranosylamide derivative (60) as oil. 1Н NMR (500 MHz, CDCl3) δ = 7.42-8.05 (m, 5H, СОC6H5), 6.30 (br.d, 1H, J1,NH = 9.3 Hz, NH), 5.11 (t, 1Н, J1,2 = J1,NH = 9.3 Hz, Н-1), 4.90 (dd, 1Н, J2,3 = 6.7 Hz, Н-2), 4.61 (dd, 1H, J6a,5 = 5.8, J6a,6́b= 11.7 Hz, H-6a), 4.53 (dd, 1H, J6b,5 = 7.2 Hz, H-6b), 4.28-4.31 (m, 3Н, Н-3, H-4, H-5), 2.12 (s, 3H, CH3CO), 1.98 (s, 3H, NHCOCH3), 1.53 and 1.35 [2s, 3H, (CH3)2C-]. 13C NMR (126 MHz, CDCl3) δ = 171.0 (NHCOCH3), 170.3 and 166.4 (2 C=O, COCH3andСОC6H5), 133.2, 129.8, 128.4 (СОC6H5), 110.8 [C-CH3)2], 76.8 (С-1), 76.2, 73.7, 72.4 (C-4, C-2, С-3, C-5), 63.7 (С-6), 27.8 and 26.2 [(CH3)2C-], 23.5 and 21.0 (COCH3andNHCOCH3). LC-MS (ESI+):m/z calcd for C20H25NO8M+Na+: 430.1, found 430.1.
Acknowledgements
This study was supported by grants from Belarusian Fond Fundamental Investigations (X-16-048) and FOI «Chemical processes, reagents and technologies, bioregulators and bioorgchemistry», s/p «Chemical foundations of life activity processes» (Bioorgchemisrty 2.3.2.2). The author expresses gratitude to D.Sc A.V. Baranovsky, Laboratory of Physicochemical Methods of Research, for writing 2D NMR spectra, and the researchers, Institute of Bioorganic Chemistry of NAN of Belarus, Belorussian State University, for their assistance in obtaining NMR, HRMS, IR spectral data, and measurement of optical rotation for carbohydrates.
References
- 1.Guimarães B, R de Oliveira Silva, V de Sales Barbosa M, Freitas J J R de, Freitas J C R de et al. (2023) . Relevant Advances in the Synthesis and Applications of N-Glycopyranosides,Chemistry Select 8, 1-13.
- 2.Sangwan R, Khanam A, Kumar Mandal P. (2020) An Overview on the Chemical N-Functionalization of Sugars and Formation of N-Glycosides,Eur. , J. Org. Chem 5949, 10-1002.
- 3.S E, M Abdur Rahman S. (2022) A Review on. Biological Activities of Sugars and Sugar Derivatives / Dhaka Univ.J. Pharm. Sci 20, 381-394.
- 4.Knauer S, Kranke B, Krause L, Kunz H. (2004) . Amino Sugars and Glycosylamines as Tools in Stereoselective Synthesis,Cur. Org. Chem.8 18, 1739-1761.
- 5.Norris P. (2008) Pyranose N-Glycosyl Amides: Emerging Targets with Diverse Biological Potential,Cur. , Top. Med. Chem 8, 101-113.
- 6.D P Gamblin, E M Scanlon, B G Davis. (2009) Glycoprotein Synthesis: An Update,Chem. , Rev 109, 131-163.
- 7.Glorgydeák Z, Hadady Z, Felfoldi N, Krakomperger A, Nagy V et al. (2004) Synthesis ofN-(β-D-glucopyranosyl)- andN-(2-acetamido-2-deoxy-β-D-glucopyranosyl) amides as inhibitors of glycogen phosphorylase,Bioorg. , Med. Chem 12, 4861-4820.
- 8.Czifrák K, Hadady Z, Docsa T, Gergely P, al Schmidt J et. (2006) Synthesis of N-(β-D-glucopyranosyl) monoamides of dicarboxylic acids as potential inhibitors of glycogen phosphorylase,Carbohydr. Res.341 947-956.
- 9.Hadjiloi T, Tiraidis C, Chrysina E, Leonidas D, Oikonomakos N et al. (2006) Binding of oxalyl derivatives of β-D-glucopyranosylamine to muscle glycogen phoshorylase,Bioorg.Med.Chem. 14, 3872-3882.
- 10.Mała P, Siebs E, Meiers J, Rox K, Varrot A et al. (2022) Discovery of N-β-L-Fucosyl Amides as High-Affinity Ligands for the Pseudomonas aeruginosa Lectin LecB,J.Med. , Chem 65, 14180-14200.
- 11.Nisic F, Bernardi A. (2011) Stereoselective synthesis of N-galactofuranosyl amides,Carbohydr. , Res 346, 465-471.
- 12.M G Traverssi, V E Manzano, Varela O, J P Colomer. (2024) Synthesis of N-glycosyl amides: conformational analysis and evaluation as inhibitors of b-galactosidase fromE. coli,RSC. Adv.14 2659-10.
- 13.Hou Z, Li C, Liu Y, Zhang M, Wang Y et al. (2020) Design, synthesis and biological evaluation of carbohydrate-based sulphonamide derivatives as topical antiglaucoma agents through selective inhibition of carbonicanhydrase II,J. , EnzymeInhib. and Med. Chem 35, 383-390.
- 14.G K Rawal, Kumar A, Tawar U, Y D Vankar. (2007) New Method for Chloroamidation of Olefins. Application in the Synthesis of N-Glycopeptides and Anticancer Agents,Org.Lett. 9, 5171-5174.
- 15.Pastuch-Gawołek G, Malarz K, Mrozek-Wilczkiewicz A, Musioła M, Serda M et al. (2016) Small molecule glycoconjugates with anticancer activity,Europ.J.Med. Chem.112 130-144.
- 16.D P Matharasi, Jayaprakash P. (2023) Synthesis, Structural, Spectral Studies and Activity Studies of 6-Methyl-4-(3-Nitro-Phenyl)-2-Oxo-1,2,3,4 Tetrahydropyrimidine-5-Carboxylic Acid Ethyl Ester Catalyzed Using BF3-OEt2,Polycyclic Aromatic Compounds. 44, 4137-4156.
- 17.L M Rosado, T J Meyerhoefer, S M Bett, Ilyas S, al Bululu L et. (2016) Direct Coupling of Amides and urea to Glycosyl Halides using Silver Triflate,Eur. , J. Org. Chem 16, 2778-2784.
- 18.Guo M, Wang X, Wang Y, Hou Z, Ch Guo et al. (2022) Facile and efficient access to C1-aminosugar derivatives under mild conditions,Tetrahedron. Lett.91 153644-10.
- 19.Pohner C, Ullmann V, Hilpert R, Samain E, Unverzagt C. (2014) Chemoselective coupling of sugar oximes and α-ketoacids to glycosyl amides and N-glycopeptides,TetrahedronLett.55. 2197-2200.
- 20.Gaitonde V, S J. (2012) Synthesis of β-Glycosyl Amides from N-Glycosyl DinitrobenzenesulfonamidesJ.Carbohydr. , Chem 31, 4-6.
- 21.Song X, R I Hollingsworth. (2006) A Stereoselective Synthesis of N-β-D-Glycosyl Amides by a Ritter-type Reaction,Synlett. 20, 3451-3454.
- 22.Nisic F, Bernardi A. (2008) Stereoselective synthesis of glycosyl amides by traceless Staudinger ligation of unprotected glycosyl azides,Carbohydr. Res.343,1636-1643.doi: 10.1016/j.caress.2008.04.023
- 23.Nisic F, Speciale G, Bernardi A. (2012) A stereoselective synthesis of α- and β-glycofuranosyl amides using the Staudinger ligation of glycofuranosyl azides,Chemistry A Eur. 18, 6895-6896.
- 24.Bianchi A, Bernardi A. (2006) Traceless Staudinger ligation of unprotected glycosyl azides with Triaryl Phosphines: Stereoselective Synthesis of glycosyl amides,J. , Org. Chem.71,4565-4577.doi.org/10.1021/jo60409s
- 25.G, A V Sivets, M A Khancheuski. (2023) Stereoselective synthesis of N-glycosyl oxazolines and evaluation of their antiproliferative activity,J. New Develop. in Chem 4, 1-23.
- 26.J D Stevens, Jr Fletcher H Gr. (1968) . The Proton Magnetic Resonance Spectra of Pentofuranose Derivatives,J. Org. Chem.33 1799-1805.
- 27.D M Gordon, S J Danishefsky. (1991) . Ritter-like Reactions of 1,3-Anhydropyranose Derivatives,J. Org. Chem.56 3713-3715.
- 28.R K Ness, H W Dieht, H G Fletcher. (1954) New Benzoyl Derivatives of D-Ribofuranosyl and aldehyde-D-Ribose. The Preparation of Crystalline 2,3,5-tri-O-benzoyl-β-D-ribose from D-ribose,J. , Am. Chem. Soc 76, 763-767.
- 29.Ochoa C, Provencio R, M L Jimeno, Balzarini J, E De Clercq. (1998) Synthesis and anti-HIV properties of 1,2,4,6-thiatriazin-3-one 1,1-dioxide nucleosides,Nucleosides. , Nucleotides 17, 901-910.
- 30.D C Lankin, S T Nugent, S N Rao. (1993) NMR spectroscopy and conformational analysis of 3-deoxy-3-C-(hyroxymethyl)-1,2;5,6-di-O-isopropylidene-α-D-allofuaranose,Carbohydr. , Res 244, 49-68.
- 31.N Savel’ ev A, F M Ibatyllin, E V, A M Kachurin, K N Neustroev. (1996) Enzymatic properties of α-D-galactosidase fromTrichodermareesei,Carbohydr. , Res 296, 261-273.