Cytoplasmic Retention of CDC6 Induces Premature Senescence in Immortalized Cells and Suppresses Tumor Formation in Mice
Abstract
Senescence is a powerful mechanism that prevents the development of tumors in vivo; however, once tumors are formed, most are refractory to senescence in response to oncogenic stress. Therefore, a novel pathway leading to senescence is required. We herein demonstrated that the cell cycle regulator CDC6 translocated from the nucleus to the cytoplasm during senescence in a leptomycin B-resistant manner. In order to evaluate the translocation of CDC6, we utilized an estrogen receptor (ER) tag to retain CDC6 in the cytoplasm. ER-tagged CDC6 was exclusively cytoplasmic, inhibited cell proliferation, and induced senescence-associated (SA) b-galactosidase activity. Furthermore, ER-CDC6 inhibited the transformation of mouse fibroblasts by the active ras oncogene in vitro, and suppressed tumor formation in NOD-SCID mice. Thus, CDC6 may play a critical role in the regulation of senescence in the cytoplasm in order to counteract tumorigenesis.
Author Contributions
Academic Editor: Fernando Luiz Affonso Fonseca, Department of Oncology
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2016 Akihiro Ueda, et al
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Mammalian cells are equipped with mechanisms that prevent the formation and development of tumors, such as checkpoint controls, DNA damage responses, differentiation, apoptosis, and senescence. Of these, senescence has been proposed to strongly suppress the formation and development of tumors1,2; however, its mechanism of action and pathways have not yet been elucidated in detail. Senescence is characterized by eternal withdrawal from the cell cycle and was originally identified as a mechanism determining the life span of primary cells cultured in vitro; however, subsequent studies reported that it was induced under several different conditions, such as telomere shortening, DNA damage, oxidative stress, the abrupt activation of oncogenes, and the developmental program3, 4, 5.
These findings demonstrated that senescence is a cellular response to stress that disables the proliferation of cells. Senescence has been recognized as a part of the system that prevents the tumorigenic transformation of mammalian cells1, 2 by inhibiting cell proliferation, and eliminates damaged cells via an inflammatory process by affecting surrounding cells through secreted factors6.
The tumor suppressor, p53, was previously shown to play an important role in senescence and tumor suppression by triggering the expression of a series of genes involved in cell cycle arrest and the onset of senescence, such as the CDK inhibitor p21, which negatively regulates G1 cyclin-CDK complexes, ultimately leading to the dephosphorylation and activation of the Rb protein3. In the initial stage of tumorigenic transformation in vivo, cells are induced to undergo senescence in order to prevent transformation7, 8, 9, 10, and p53 has been shown to play a role in this process11, 12. However, when cells go through this anti-tumorigenic barrier, tumor cells acquire mutations in relevant genes such as those involved in the p53 pathway, and become refractory to senescence. Although previous studies reported that a p53-independent mechanism was involved in the control of senescence13, 14, 15, 16, 17 and also that Rb, but not other members of the Rb family, played an essential role in senescence18, novel molecular switches to initiate the senescence program, which are desired to counteract tumorigenic transformation, have not yet been identified.
During the course of our research on the COP9 signalosome (CSN) complex19, 20, an upstream regulator of the cullin-RING ubiquitin ligase (CRL) complex, we found that gene-knockout of the fifth component of CSN (CSN5) induced premature senescence in mouse fibroblasts, even in the absence of p5321. The depletion of CSN5 increased CDK2 and cyclin E levels in the cytoplasm, whereas the concomitant knockdown of CDK2 reduced cytoplasmic cyclin E levels and rescued the induction of senescence22. Cytoplasmic cyclin E was previously detected in mouse embryonic fibroblasts (MEFs) that entered proliferative senescence22. The enforced expression of cytoplasmic cyclin E in immortalized mouse and human cells has been shown to induce senescence22. Thus, the cytoplasmic retention of cyclin E, CDK2, and their interacting proteins was identified as a key event that induced premature senescence in a novel manner.
In the present study, we focused on the cell cycle regulator CDC6 as one of the interactors of cyclin E and CDK223. CDC6 is known to play an essential role in the initiation of DNA replication24. Recent findings demonstrated that CDC6 was crucially involved in cell cycle checkpoints and oncogenesis25. However, these functions are largely executed in the nucleus. We detected CDC6 in the cytoplasm of senescent cells in a leptomycin B-resistant manner. In order to examine the physiological significance of cytoplasmic CDC6, we ectopically expressed CDC6 that was largely retained in the cytoplasm, which inhibited proliferation and induced premature senescence. Furthermore, cytoplasmic CDC6 inhibited tumor formation by mouse fibroblasts transformed by the activated ras oncogene, indicating that the cytoplasmic retention of CDC6 prevented tumorigenesis through the induction of premature senescence in a novel manner.
Materials and Methods
Cell Culture and Transfection.
MEFs harboring null (-) and conditional (floxed, f) alleles of the CSN5/Jab1 gene and immortalized by the null allele of p53 were described previously21, 22. MEFs, NIH3T3 (Arf-null, p53-wild-type) mouse fibroblasts (provided by Drs C. J. Sherr and M. F. Roussel), and 293T human embryonic kidney (HEK) cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2mM glutamine, 100 units/ml of penicillin, and 100 μg/ml of streptomycin (GIBCO/BRL). Transfection with expression vectors was performed using the calcium phosphate-DNA precipitation method42. In the focus formation assay, transfected cells were maintained in medium containing 5% FBS instead of 10% for approximately 2-3 weeks. In the colony formation assay, transfected cells were selected for resistance to puromycin (5 mg/ml) or G418 (1 mg/ml), depending on the type of vector used for transfection for 2-4 weeks. Both foci and colonies were fixed in 4% paraformaldehyde, and stained in 0.4% crystal violet. In some cases, selected cells were sorted for GFP-positive signals with a FACS Aria flow cytometer (Becton Dickinson).
Plasmid Construction
A cDNA fragment containing the entire coding sequences of mouse CDC6 with or without fusion to the sequence of the estrogen receptor (ER)29 was inserted into the HA-tagged expression vector (a gift from Dr. Junichi Fujisawa).
Protein Analyses
Cell lysis, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and immunoblotting were performed as described previously43, 44. A mouse monoclonal antibody to CDC6 (180.2) and rabbit polyclonal antibody to cyclin E (M20) were obtained from Santa Cruz. Mouse monoclonal antibodies to g-tubulin (GTU88) and an HA epitope (12CA5) were purchased from Sigma and Boehringer Mannheim, respectively.
Immunofluorescence Staining
Cells grown on coverslips were fixed in 4% paraformaldehyde, permeabilized in 0.5% Triton X-100, stained with rabbit polyclonal and mouse monoclonal antibodies, and incubated with Alexa Fluor 594-conjugated anti-mouse/rabbit IgG (Invitrogen). Chromosomal DNA was stained by a 2-min incubation in 1 mg/ml of Hoechst 33342. Cell samples were viewed using phase contrast or fluorescence microscopy to enumerate fluorescence-positive cells.
In Vitro Binding Assay
A cDNA fragment containing the entire coding sequence of mouse cyclin E was inserted into the pGEX vector (Pharmacia) in-frame with glutathione S-transferase (GST). The expression and purification of GST-fused proteins and the binding conditions used were as described previously44.
b-Galactosidase Assay
Cells were fixed in 0.25% glutaraldehyde, incubated for 16-24 hours in X-Gal solution containing 0.2% X-Gal, 2 mM MgCl2, 5 mM K4Fe(CN)6, and 5 mM K3Fe(CN)6, and viewed with a phase contrast microscope.
Tumorigenicity Assay
Cells (ca 106) were subcutaneously injected into 6-week-old NOD-SCID mice. Mice were sacrificed and the sizes of tumors were measured 2-3 weeks post-injection. All mice were maintained in the Nara Institute of Science and Technology Animal Facility in accordance with Nara Institute of Science and Technology guidelines. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Animal Research Management Committee of Nara Institute of Science and Technology (Permit Number: 1310). All surgery was performed under isoflurane anesthesia, and all efforts were made to minimize suffering.
Results
CDC6 was located in the Cytoplasm of Senescent Cells.
We previously reported that knockout of the CSN5 gene induced premature senescence in mouse fibroblasts, even in the absence of p5321. We found that cyclin E levels were higher in the cytoplasm of these cells22, suggesting that certain cyclin E-interacting proteins are located in the cytoplasm of CSN5-knockout cells and may trigger the senescence-initiating program. We suspected that CDC6 was one such protein and subsequently investigated this possibility. CDC6 has been shown to play an important role in the regulation of DNA replication by assisting in the assembly of the pre-replication complex (RC) at the G1 phase of the cell cycle24. In order to achieve this, CDC6 exerts its effects in the nucleus with the aim of associating with origin recognition complex (ORC) proteins. After the onset of the S phase, CDC6 is phosphorylated, ubiquitinated, translocated to the cytoplasm, and degraded by the proteasome to avoid re-replication26, 27, 28. If CDC6 plays a role in senescence and is involved in the cytoplasmic location of cyclin E, it may be located in the cytoplasm of senescent cells.
CDC6 was largely located in the nuclei of proliferating NIH3T3 cells and MEFs harboring p53-/- alleles, which are immortal and do not enter senescence, and cytoplasmic staining was weak (Figure 1a, b). This result was consistent with previous findings in which the CDC6 protein exported from the nucleus in proliferating cells was unstable and rapidly degraded through the ubiquitin-proteasome system26, 27, 28However, the CDC6-specific fluorescent signal was significantly strong in the cytoplasm of CSN5-null (CSN5-/D) MEFs, which were generated by introducing CRE-ER into CSN5-/floxed p53-/- MEFs followed by a treatment with 4OHT (Figure 1a). Approximately 40% of proliferating MEFs contained nuclear CDC6, whereas less than 5% of senescent MEFs showed nucleus-specific CDC6 staining (Figure 1b).
We utilized replicative-senescent cells to determine whether cytoplasmic CDC6 was restricted to CSN5-knockdown cells. Wild-type MEF cells were cultured in vitro, and the subcellular localization of CDC6 was compared between early (less than 5 passages) and late (more than 10 passages) passages (Figure 1c, d). Proliferating pre-senescent MEF cells (early passage) exhibited similar staining patterns to NIH3T3 cells. However, nuclear staining was not strong in late passage MEFs and the CDC6-specific fluorescent signal was significantly strong in the cytoplasm (Figure 1c). Approximately 50% of proliferating MEFs contained nuclear CDC6, whereas less than 5% of senescent MEFs showed clear nuclear CDC6 staining (Figure 1d). Furthermore, cytoplasmic staining of CDC6 in late passage MEFs was resistant to leptomycin B (LMB), a chemical inhibitor to nuclear export (Figure 1c, d), which indicated that the cytoplasmic location of CDC6 was not due to accelerated nuclear export, but rather to cytoplasmic retention.
Figure 1.Subcellular localization of endogenous CDC6 in mouse fibroblasts before and after entering senescence. (a, b) Proliferating mouse NIH3T3 cells, and CSN5-/floxed MEFs before (CSN5-/floxed) and after (CSN5-/D) the treatment with CRE were fixed and stained with an antibody to mouse CDC6. Chromosomal DNA was visualized by staining with Hoechst 33342. (c, d) Wild-type MEFs cultured in vitro for less than 5 passages (early passage) and more than 10 passages (late passage) treated with (+) and without (-) leptomycin B (LMB) were fixed and stained as in panel a. Photos of phase contrast (PC) and fluorescence microscopy (DNA and aCDC6) (a, c) and the results of enumeration (b, d) are shown.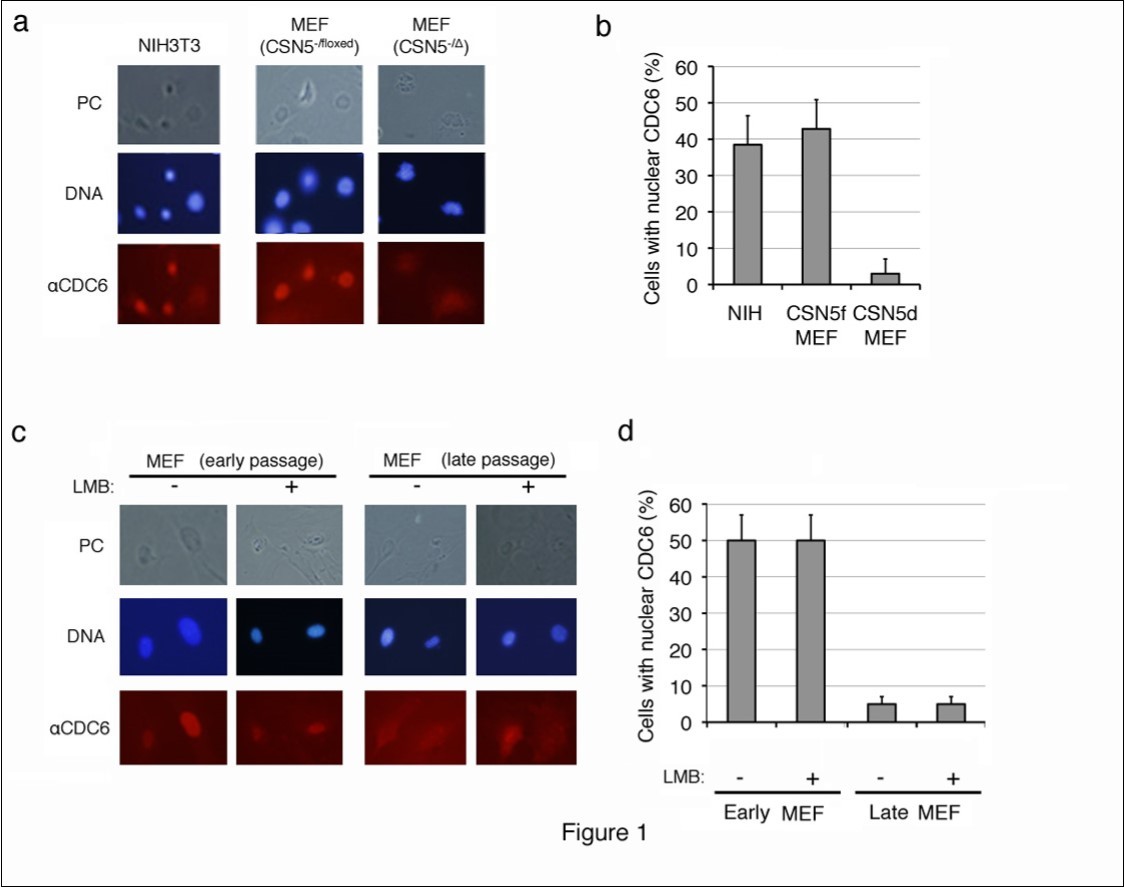
ER-Fused CDC6 was Predominantly located in the Cytoplasm, Inhibited Cell Proliferation, and Induced Senescence.
In order to explore the possible roles of cytoplasmic CDC6, CDC6 was predominantly expressed in the cytoplasm. Since cytoplasmic CDC6 in senescent cells was refractory to the effects of LMB, we enforced the cytoplasmic retention of CDC6 rather than enhancing its export from the nucleus, and fused CDC6 with an ER tag to achieve this29. ER is a nuclear receptor that is exclusively retained in the cytoplasm in the absence of a ligand. Therefore, ER tagging is a very useful method for controlling the intracellular localization of a fused protein. We constructed an expression vector encoding HA-tagged CDC6 (HA-CDC6) or CDC6 tagged with both HA and ER (HA-CDC6-ER), and transfected 293T cells with these vectors. The cell lysates isolated from the transfectants were analyzed by immunoblotting with an antibody to an HA epitope. Figure 2a shows that HA-CDC6 and HA-CDC6-ER proteins were both detected without significant degradation. We then transfected NIH3T3 cells with a control vector and expression vector encoding HA-CDC6 or HA-CDC6-ER together with a marker plasmid containing green fluorescent protein (GFP) cDNA and a neo-resistant gene. Transiently transfected cells were fixed and stained with an antibody to the HA epitope. Figure 2b shows that HA-CDC6 was predominantly located in the nucleus, whereas HA-CDC6-ER was largely located in the cytoplasm.
Figure 2.Localization of ER-tagged CDC6. (a) HEK293T cells were transfected with a mammalian expression vector containing HA-fused CDC6 with (HA-CDC6-ER) and without (HA-CDC6) an ER tag. Lysates isolated from transfected cells were analyzed by Western blotting using an antibody specifically recognizing an HA epitope. (b) NIH3T3 cells transfected with expression vectors (HA-CDC6 and HA-CDC6-ER) together with a GFP marker plasmid were fixed and stained with an antibody specific to an HA epitope. Cells transfected with HA-CDC6-ER were incubated in the presence (+4OHT) and absence (-4OHT) of 4OHT for 2 additional days and then fixed and stained as above. Chromosomal DNA was visualized by staining with Hoechst 33342.
Since ER is transferred to the nucleus after binding to its ligand (ex., 4-hydroxytamoxifen (4OHT)), we incubated cells transfected with HA-CDC6-ER in the presence and absence of 4OHT for 2 additional days (Figure 2b). The nuclear signal of CDC6 was stronger in the presence than in the absence of 4OHT; however, a large amount of CDC6 still remained in the cytoplasm, which demonstrated that ligand binding transferred some, but not all ER-fused proteins into the nucleus in this system.
We performed a colony-formation assay in order to examine the effects of cytoplasmic CDC6 on cell proliferation. We transfected NIH3T3 cells with HA-CDC6 and HA-CDC6-ER vectors together with a GFP-neo marker plasmid, and selected cells in the presence of G418. After 2 weeks, cells transfected with the HA-CDC6 vector formed as many colonies as those with an empty vector control, whereas HA-CDD6-ER-transfectants formed none (Figure 3a). The incubation of cells transfected with HA-CDC6-ER and a GFP-neo marker in the presence of 4OHT from the beginning of transfection failed to rescue the formation of colonies. These results indicated that cytoplasmic CDC6 dominated the effects of nuclear CDC6. In order to test this hypothesis, we transfected cells with a constant amount of HA-CDC6-ER together with increasing amounts of HA-CDC6 and allowed them to form colonies. The results shown in Figure 3b demonstrated that nuclear CDC6 did not override the inhibitory effects of cytoplasmic CDC6. Thus, it was not the disappearance of CDC6 from the nucleus, but the cytoplasmic localization of CDC6 that induced the inhibition of proliferation.
Figure 3.Colony formation assay with CDC6 expression vectors. (a) NIH3T3 cells were transfected with HA-CDC6 and HA-CDC6-ER vectors together with a GFP-neo marker plasmid, and were selected in G418 for 2 weeks. HA-CDC6-ER-transfectants were incubated in the presence of 4OHT (+4OHT) from the beginning of transfection and selected in G418. (b) NIH3T3 cells were transfected with a constant amount of the HA-CDC6-ER (1mg) vector together with a GFP-neo marker plasmid, and also with an increasing amount of the HA-CDC6 vector, and then selected in G418. Colonies were fixed and stained with 0.1% crystal violet.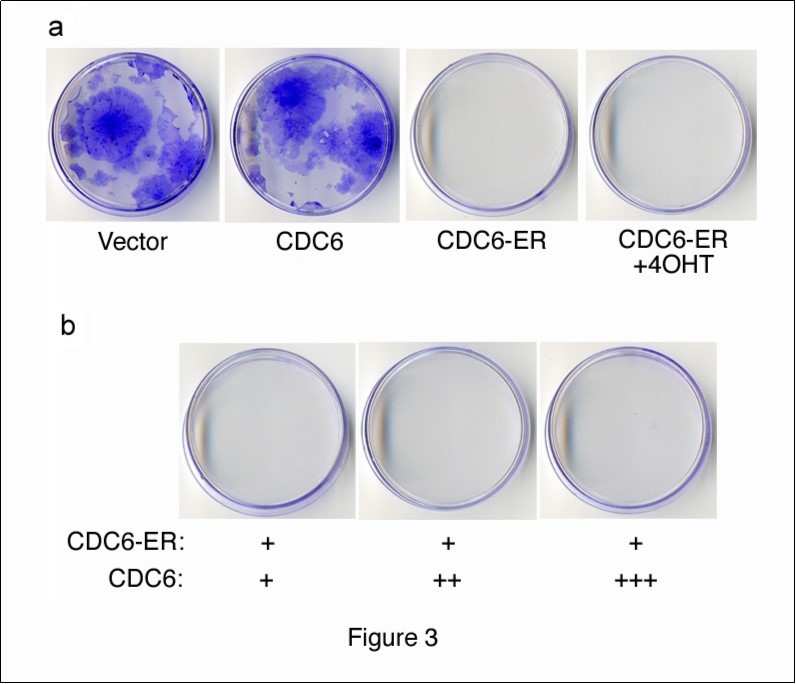
Although no colonies were formed, significant number of cells remained attached to the plate transfected with HA-CDC6-ER followed by selection in G418, and we performed an assay to measure senescence-associated-beta-galactosidase (SA-b-gal) activity. Figure 4 shows that transfection with HA-CDC6-ER induced a significant number of SA-b-gal-positive cells, while that with HA-CDC6 or a control vector did not. As expected, incubation in the presence of 4OHT from the beginning of transfection failed to suppress the induction of SA-b-gal activity in cells transfected with HA-CDC6-ER.
Figure 4.Cytoplasmic CDC6 induced SA-b-Gal activity NIH3T3 cells were transfected with HA-CDC6 and HA-CDC6-ER vectors together with a GFP-neo marker plasmid, selected in G418, and stained for SA-b-Gal activity. HA-CDC6-ER-transfectants were incubated in the presence of 4OHT (+4OHT) from the beginning of transfection, selected in G418, and stained. Photos of stained cells (a) and the results of enumeration (b) are shown.
ER tagging is a widely used method to control the subcellular distribution of a protein between the nucleus and cytoplasm with few side effects. For example, an ER-tagged CRE recombinase is generally used to induce conditional knockout in mice and cells containing floxed sequences in the loci of interest. We previously used the CRE-ER system to conditionally knockout the CSN5 allele without any undesirable side effects 22, 30, and successfully applied this system to other proteins22. Therefore, the results obtained with ER-tagged CDC6 were attributed to the cytoplasmic localization of the CDC6 protein, but not to the expression of ER.
Taken together, although cells expressing cytoplasmic CDC6 remained alive (Figure 4), their proliferation was inhibited (Figure 3), and a senescence marker was induced (Figure 4). Furthermore, an increase in the amount of nuclear CDC6 did not overcome the cytoplasmic CDC6-mediated inhibition of proliferation (Figure 3). Therefore, we concluded that the cytoplasmic localization of CDC6 triggered the senescence-initiation program.
CDC6 Bound to Cyclin E in Vitro and in Vivo.
CDC6 is a well-known substrate of CDK2 kinase28, and is expected to interact with cyclin E and CDK223. We confirmed the interaction between CDC6 and cyclin E using an in vitro binding assay. Recombinant GST and GST-fused cyclin E proteins produced in bacteria were immobilized on glutathione beads and mixed with a cell lysate containing the ectopically-expressed HA-tagged CDC6 protein. Bound CDC6 was detected by immunoblotting using an antibody to an HA epitope.
a showed that CDC6 interacted with cyclin E in vitro.
We next determined whether cytoplasmic CDC6 interacted with cyclin E in cells. We transfected NIH3T3 cells with HA-CDC6 and HA-CDC6-ER vectors and harvested 4 days later. HA-CDC6 and HA-CDC6-ER proteins were collected from each lysate by immunoprecipitation with an antibody to an HA epitope, and the bound cyclin E protein was detected by immunoblotting using a specific antibody to cyclin E. Figure 5b shows that nuclear (HA-CDC6) and cytoplasmic (HA-CDC6-ER) CDC6 proteins both equivalently bound to cyclin E in cells.
Figure 5.Cytoplasmic CDC6 interacted with and translocated cyclin E to the cytoplasm. (a) GST and GST-Cyclin E (CycE) proteins produced in E. coli and immobilized on glutathione beads were incubated with crude cell extracts containing the HA-tagged CDC6 protein, separated by SDS-PAGE, and visualized by CBB staining (lower panel). The bound protein was detected by immunoblotting with an antibody to an HA epitope (upper panel). (b) NIH3T3 cells were transfected with HA-CDC6 and HA-CDC6-ER vectors and were harvested 4 days later. HA-CDC6 and HA-CDC6-ER proteins contained in the lysate were collected by immunoprecipitation with an antibody to an HA epitope, and the bound protein was detected by immunoblotting using antibodies to an HA epitope and cyclin E. NRS; normal rabbit serum, Ig; immunoglobulin. (c, d) NIH3T3 cells transfected with HA-CDC6 and HA-CDC6-ER vectors together with a GFP-neo marker plasmid were fixed and stained with an antibody specific to cyclin E. Chromosomal DNA was visualized by staining with Hoechst 33342. Photos of phase contrast (PC) and fluorescence microscopy (DNA, GFP, and aCycE) (c) and the results of enumeration (d) are shown.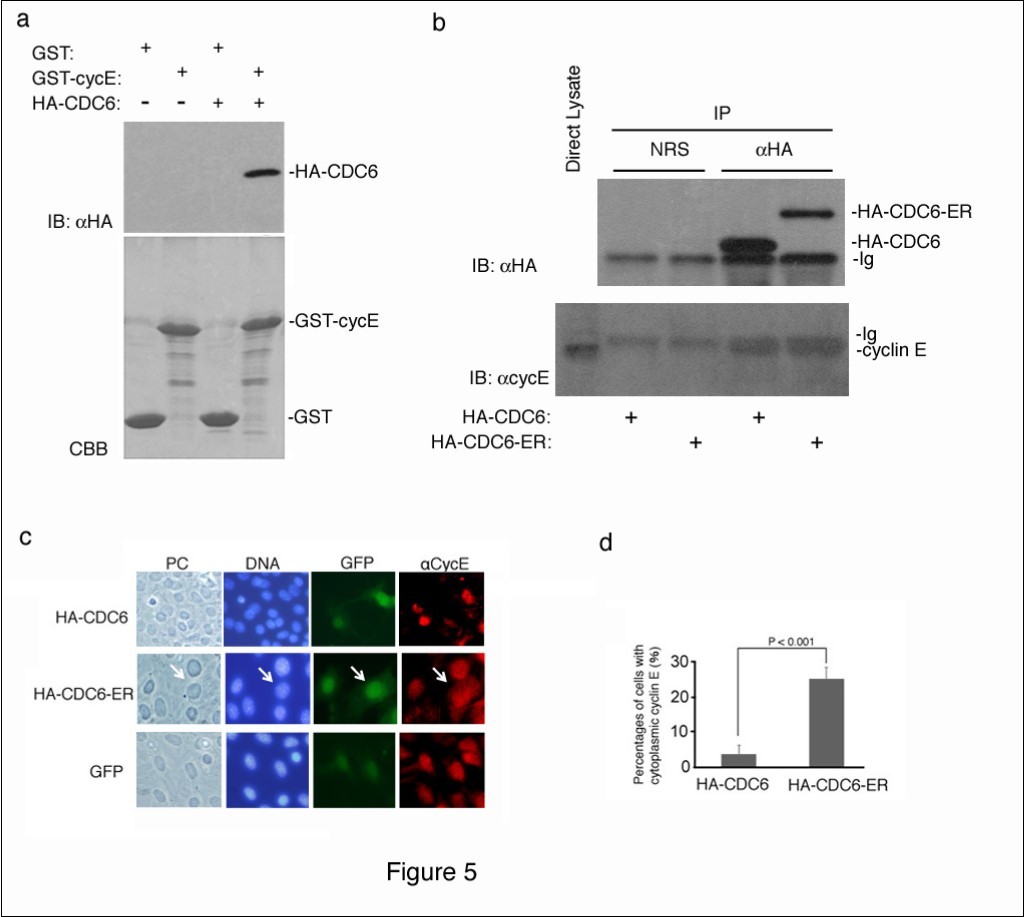
Cyclin E Translocated to the Cytoplasm in CDC6-ER-Transfected Cells.
Cyclin E was detected in the cytoplasm of CSN5-null cells that underwent senescence, and the enforced expression of cyclin E in the cytoplasm induced senescence22. Since CDC6 interacted with cyclin E in vitro and in vivoFigure 5a, b), we investigated the intracellular localization of cyclin E in cells transfected with HA-CDC6/HA-CDC6-ER by immunofluorescent staining with a specific antibody to cyclin E. Figure 5c shows that cyclin E was exclusively located in the nucleus in some cells that were introduced with GFP and HA-CDC6. In contrast, a significant percentage of HA-CDC6-ER-transfected cells had cytoplasmic cyclin E in addition to nuclear cyclin E (Figure 5c). A quantitative analysis revealed that this difference was significant (Figure 5d), indicating that the cytoplasmic expression of CDC6 converted a certain amount of cyclin E from the nucleus to the cytoplasm, and may have initiated a senescence program together with CDC6.
Cytoplasmic CDC6 Inhibited Transformation Mediated by Active Ras.
We next investigated whether cytoplasmic CDC6 prevented transformation by an active oncogene in vitro and in vivo. The introduction of the active form of Ras is known to transform immortalized mouse fibroblasts such as NIH3T3 cells, and makes them lose contact inhibition and anchorage-dependent growth, thereby allowing the formation of foci on a monolayer of cells. We utilized this assay to evaluate the anti-tumorigenic activity of cytoplasmic CDC6. We transfected NIH3T3 cells with an expression vector containing the active form of Ha-Ras together with an HA-CDC6 or HA-CDC6-ER vector, maintained the culture for 2 weeks, and then performed staining with crystal violet (Figure 6a, b). The introduction of Ha-ras into NIH3T3 cells induced the formation of foci as expected, and co-transfection with HA-CDC6 did not significantly affect the number or size of foci induced by Ha-ras. In contrast, the co-introduction with HA-CDC6-ER markedly inhibited the formation of foci.
In order to examine the effects of cytoplasmic CDC6 on tumorigenesis in more detail, we transfected NIH3T3 cells with Ha-ras and HA-CDC6/HA-CDC6-ER together with a GFP vector, collected successful transfectants using the GFP signal with a cell sorter, and subcutaneously injected them into NOD-SCID mice (Figure 6c, d). After 2-3 weeks, control NIH3T3 cells (GFP) failed to proliferate within the bodies of mice, whereas cells transformed by ras formed large tumors in all mice injected. Co-transfection with HA-CDC6 did not significantly affect the efficiency of tumorigenesis, size of tumors, or time kinetics of developing tumors. In contrast, the co-introduction of HA-CDC6-ER markedly impaired the formation of tumors. Although the formation of ras-mediated tumors was often observed 1 week after the injection, no trace of tumor cells was detected 4 weeks after the injection of CDC6-ER-co-transfected cells into mice. Thus, we concluded that the cytoplasmic localization of CDC6 prevented the transformation of mouse fibroblasts by the activated ras oncogene.
Figure 6.Cytoplasmic CDC6 suppressed focus and tumor formation by cells transformed using the activated ras oncogene. (a, b) NIH3T3 cells were transfected with a vector containing the active form of Ha-ras, together with HA-CDC6 and HA-CDC6-ER vectors. The resulting foci were fixed and stained with 0.1% crystal violet 2-3 weeks post-transfection. Photos of actual foci (a) and the results of enumeration (b) are shown. (c, d) NIH3T3 cells were transfected with active Ha-Ras and HA-CDC6/HA-CDC6-ER together with a GFP-neo marker plasmid. GFP-positive cells were enriched with a cell sorter and subcutaneously injected into NOD-SCID mice. After 2-3 weeks, mice were sacrificed and the sizes of tumors were measured. Photos of mice (a) and the results of measurements of tumors (b) are shown.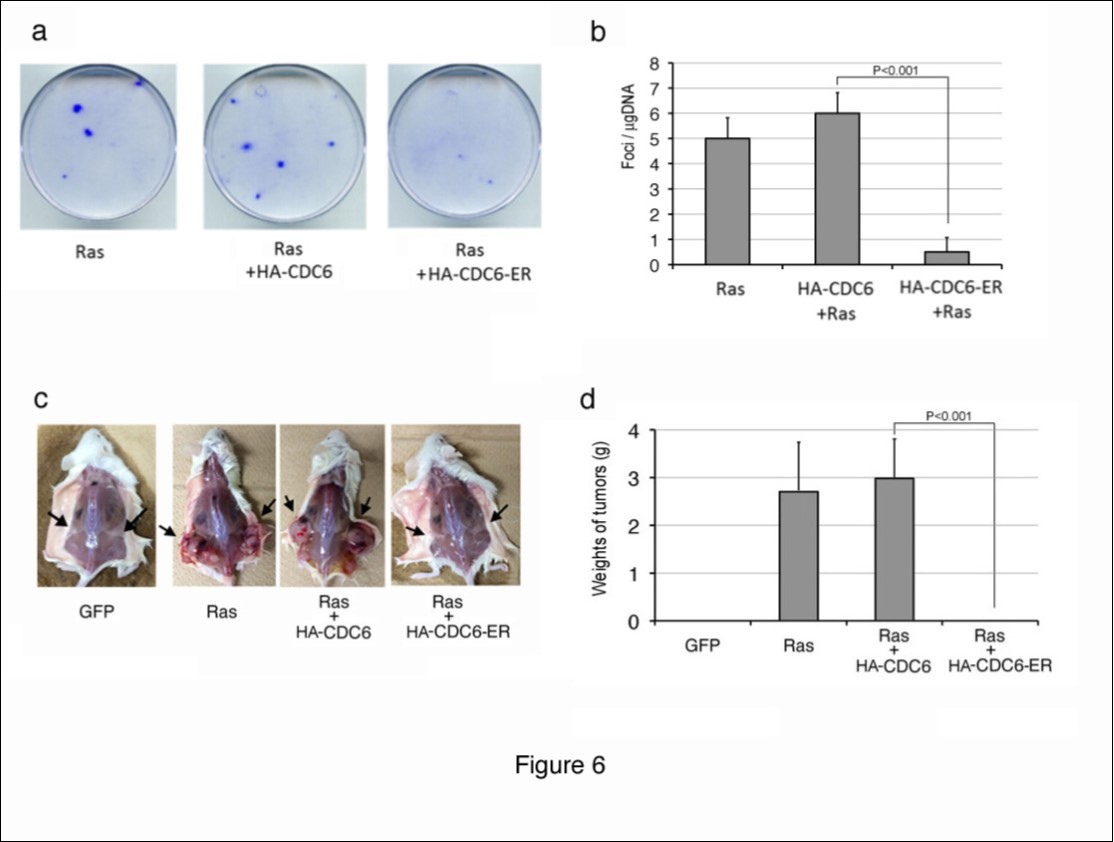
Discussion
The function of the CSN is critical to mammalian cell proliferation and tumorigenesis, and the expression of the fifth subunit was previously shown to frequently be elevated in many human cancers20, indicating that CSN5 possesses a specific function that is closely connected to tumor cell proliferation. We previously reported that CSN5, but not the whole CSN complex, interacted with a key regulatory element of the cell cycle, CDK222. The interaction between CSN5 and CDK2 participated in the control of premature senescence, and, in this process, CDK2 and its partner cyclin E were retained in the cytoplasm. The enforced expression of cyclin E in the cytoplasm induced senescence22, suggesting that cyclin E has a novel function in the cytoplasm to induce senescence. In the present study, we focused on CDC6 because it is a key molecule involved in cell cycle regulation and is also an interactor of cyclin E. We found that CDC6 was located in the cytoplasm of senescent cells induced by a late passage and CSN5 knockout, whereas it was compartmentalized into the nucleus in proliferating cells. The cytoplasmic location of CDC6 in senescent cells was refractory to the effects of LMB, suggesting that cytoplasmic retention rather than enhanced nuclear export was responsible for this phenomenon. The enforced expression of CDC6 in the cytoplasm induced senescence, similar to that by cytoplasmic cyclin E, suggesting that CDC6 may also possess a novel function that ultimately leads to senescence. Cyclin E translocated to the cytoplasm of cells in which the expression of CDC6 was enforced in the cytoplasm, suggesting that cyclin E and CDC6 may have cooperated with each other to induce senescence.
CDC6 plays a key role in DNA replication and cell cycle progression; therefore, it is expected to have oncogenic properties. The CDC6 gene was previously reported to be amplified and overexpressed in human gastric cancer31. High levels of CDC6 have also been detected in brain tumors32, non-small cell lung carcinomas33, and mantle cell lymphomas34. However, the overexpression of CDC6 does not necessarily correlate with increased proliferation33, suggesting that CDC6 has oncogenic activities other than accelerated DNA replication. Previous studies reported that deregulated CDC6 inactivated the INK4A/ARF locus, which encodes tumor suppressor proteins that function upstream of the Rb and p53 pathways35, that the expression of CDC6 repressed E-cadherin transcription36, and also that CDC6 obstructed apoptosome assembly by binding to Apaf-137. In the present study, we showed that cytoplasmic CDC6, which was exclusively observed in senescent cells, exerted anti-tumorigenic activity, possibly as a result of interference with the oncogenic pathway in which nuclear CDC6 participates. Among the possible functions of CDC6 described above, p53 is known to be connected with the senescent pathway18; however, the mouse fibroblast cells used in the present study lacked p53 and INK4A/ARF loci, suggesting that cytoplasmic CDC6 did not act through this pathway, and that CDC6 may trigger a novel pathway to initiate the senescence program in the cytoplasm.
Novel factors and pathways leading to senescence (or protecting cells from senescence) have recently been reported. Palbociclib (PD-0332991), a selective inhibitor of cyclin D-dependent kinases (CDK4 and CDK6)39, has been shown to induce senescence in several human cancer cells, including breast cancer cells, and is now being developed as a drug for the treatment of ER-positive and HER2-negative breast cancer40, which implies that cyclin D-dependent kinases suppress senescence in this type of cancer. SPH2 is a tyrosine phosphatase, and its pharmacological inhibition has been shown to induce senescence in breast cancer cells by upregulating the activities of p27 and p5338. MT1-MMP (MMP14), a collagenolytic enzyme, has been implicated in extracellular matrix (ECM) remodeling, with its loss inducing senescence, which may be partially rescued using all-trans retinoic acid41. These findings indicate that as-yet unidentified pathways leading to senescence require further study. We herein revealed a novel pathway through which the cytoplasmic retention of CDC6 induced senescence. Cytoplasmic CDC6 may have interfered with the oncogenic pathway in which nuclear CDC6 participates, or CDC6 and cyclin E may have initiated a novel senescence program in the cytoplasm. Whatever the mechanism, identifying small molecules that induce the cytoplasmic retention of CDC6 and/or cyclin E may present a novel approach to developing a new type of anti-cancer drug that will induce senescence in tumor cells.
Acknowledgments
We thank Drs. CJ Sherr and MF Roussel for the NIH3T3 cell line, Dr. J Fujisawa for the HA-tagged expression vector, and Drs. Ruggero Pardi and Martina Panattoni for the CSN5-floxed mice. This work was supported by Grants-in-Aid for Scientific Research and for Cancer Research from the Ministry of Education, Science, and Culture of Japan.
References
- 1.Acosta J C, Gil J. (2012) Senescence: a new weapon for cancer therapy. , Trends Cell Biol 22, 211-219.
- 2.Collado M, Serrano M. (2010) Senescence in tumours: evidence from mice and humans. , Nat Rev Cancer 10, 51-57.
- 3.Munoz-Espin D, Serrano M. (2014) Cellular senescence: from physiology to pathology. , Nat Rev Mol Cell Biol 15, 482-496.
- 4.Yaswen P, Campisi J. (2007) Oncogene-induced senescence pathways weave an intricate tapestry. , Cell 128, 233-234.
- 5.Salama R, Sadaie M, Hoare M, Narita M. (2014) Cellular senescence and its effector programs. , Genes Dev 28, 99-114.
- 6.Kuilman T, Michaloglou C, Mooi W J, Peeper D S. (2010) The essence of senescence. , Genes Dev 24, 2463-2479.
- 7.Chen Z, Trotman L C, Shaffer D, Lin H K, Dotan Z A. (2005) Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. , Nature 436, 725-730.
- 8.Michaloglou C, Vredeveld L C, Soengas M S, Denoyelle C, Kuilman T. (2005) BRAFE600-associated senescence-like cell cycle arrest of human naevi. , Nature 436, 720-724.
- 9.Braig M, Lee S, Loddenkemper C, Rudolph C, Peters A H. (2005) Oncogene-induced senescence as an initial barrier in lymphoma development. , Nature 436, 660-665.
- 10.Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher A J. (2005) Tumour biology: senescence in premalignant tumours. , Nature 436, 642.
- 11.Meek D W. (2009) Tumour suppression by p53: a role for the DNA damage response?. , Nat Rev Cancer 9, 714-723.
- 12.Zilfou J T, Lowe S W. (2009) Tumor suppressive functions of p53. , Cold Spring Harb Perspect Biol 1, 001883.
- 13.Ha L, Ichikawa T, Anver M, Dickins R, Lowe S. (2007) ARF functions as a melanoma tumor suppressor by inducing p53-independent senescence. , Proc Natl Acad Sci U S A 104, 10968-10973.
- 14.Jacobs J J, T de Lange. (2004) Significant role for p16INK4a in p53-independent telomere-directed senescence. , Curr Biol 14, 2302-2308.
- 15.Lin H K, Chen Z, Wang G, Nardella C, Lee S W. (2010) Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. , Nature 464, 374-379.
- 16.Zou X, Ray D, Aziyu A, Christov K, Boiko A D. (2002) Cdk4 disruption renders primary mouse cells resistant to oncogenic transformation, leading to Arf/p53-independent senescence. , Genes Dev 16, 2923-2934.
- 17.Xu M, Yu Q, Subrahmanyam R, Difilippantonio M J, Ried T. (2008) Beta-catenin expression results in p53-independent DNA damage and oncogene-induced senescence in prelymphomagenic thymocytes in vivo. , Mol Cell Biol 28, 1713-1723.
- 18.Chicas A, Wang X, Zhang C, McCurrach M, Zhao Z. (2010) Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. , Cancer Cell 17, 376-387.
- 21.Yoshida A, Yoneda-Kato N, Panattoni M, Pardi R, Kato J Y. (2010) CSN5/Jab1 controls multiple events in the mammalian cell cycle. , FEBS Lett 584, 4545-4552.
- 22.Yoshida A, Yoneda-Kato N, Kato J Y. (2013) CSN5 specifically interacts with CDK2 and controls senescence in a cytoplasmic cyclin E-mediated manner. , Sci Rep 3, 1054.
- 23.Furstenthal L, Kaiser B K, Swanson C, Jackson P K. (2001) Cyclin E uses Cdc6 as a chromatin-associated receptor required for DNA replication. , J Cell Biol 152, 1267-1278.
- 25.Borlado L R, Mendez J. (2008) CDC6: from DNA replication to cell cycle checkpoints and oncogenesis. , Carcinogenesis 29, 237-243.
- 26.Petersen B O, Wagener C, Marinoni F, Kramer E R, Melixetian M. (2000) Cell cycle- and cell growth-regulated proteolysis of mammalian CDC6 is dependent on APC-CDH1. , Genes Dev 14, 2330-2343.
- 27.Mailand N, Diffley J F. (2005) CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. , Cell 122, 915-926.
- 28.Petersen B O, Lukas J, Sorensen C S, Bartek J, Helin K. (1999) Phosphorylation of mammalian CDC6 by cyclin A/CDK2 regulates its subcellular localization. , EMBO J 18, 396-410.
- 29.Matsuda T.Cepko CL (2007) Controlled expression of transgenes introduced by in vivo electroporation. , Proc Natl Acad Sci U S A 104, 1027-1032.
- 30.Tsujimoto I, Yoshida A, Yoneda-Kato N, Kato J Y. (2012) Depletion of CSN5 inhibits Ras-mediated tumorigenesis by inducing premature senescence in p53-null cells. , FEBS Lett 586, 4326-4331.
- 31.Santarius T, Shipley J, Brewer D, Stratton M R, Cooper C S. (2010) A census of amplified and overexpressed human cancer genes. , Nat Rev Cancer 10, 59-64.
- 32.Ohta S, Koide M, Tokuyama T, Yokota N, Nishizawa S. (2001) Cdc6 expression as a marker of proliferative activity in brain tumors. , Oncol Rep 8, 1063-1066.
- 33.Karakaidos P, Taraviras S, Vassiliou L V, Zacharatos P, Kastrinakis N G. (2004) Overexpression of the replication licensing regulators hCdt1 and hCdc6 characterizes a subset of non-small-cell lung carcinomas: synergistic effect with mutant p53 on tumor growth and chromosomal instability--evidence of E2F-1 transcriptional control over hCdt1. , Am J Pathol 165, 1351-1365.
- 34.Pinyol M, Salaverria I, Bea S, Fernandez V, Colomo L. (2006) Unbalanced expression of licensing DNA replication factors occurs in a subset of mantle cell lymphomas with genomic instability. , Int J Cancer 119, 2768-2774.
- 35.Gonzalez S, Klatt P, Delgado S, Conde E, Lopez-Rios F. (2006) Oncogenic activity of Cdc6 through repression of the INK4/ARF locus. , Nature 440, 702-706.
- 36.Sideridou M, Zakopoulou R, Evangelou K, Liontos M, Kotsinas A. (2011) Cdc6 expression represses E-cadherin transcription and activates adjacent replication origins. , J Cell Biol 195, 1123-1140.
- 37.Niimi S, Arakawa-Takeuchi S, Uranbileg B, Park J H, Jinno S. (2012) Cdc6 protein obstructs apoptosome assembly and consequent cell death by forming stable complexes with activated Apaf-1 molecules. , J Biol Chem 287, 18573-18583.
- 38.Lan L, Holland J D, Qi J, Grosskopf S, Vogel R. (2015) Shp2 signaling suppresses senescence in PyMT-induced mammary gland cancer in mice. , EMBO J 34, 1493-1508.
- 39.Fry D W, Harvey P J, Keller P R, Elliott W L, Meade M. (2004) Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. , Mol Cancer Ther 3, 1427-1438.
- 40.Finn R S, Dering J, Conklin D, Kalous O, Cohen D J. (2009) PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res 11:R77.
- 41.Gutierrez-Fernandez A, Soria-Valles C, Osorio F G, Gutierrez-Abril J, Garabaya C. (2015) Loss of MT1-MMP causes cell senescence and nuclear defects which can be reversed by retinoic acid. , EMBO J 34, 1875-1888.
- 42.Chen C, Okayama H. (1987) High-efficiency transformation of mammalian cells by plasmid DNA. , Mol Cell Biol 7, 2745-2752.
Cited by (2)
This article has been cited by 2 scholarly works according to:
Citing Articles:
International Journal of Cancer (2020) OpenAlex
Nicho Lim, P. Townsend - International Journal of Cancer (2020) Semantic Scholar
International Journal of Cancer (2020) Crossref