The Daughter of Time: Late Development of Waldenstrom’s Macroglobulinemiain a Patient with Immunotactoid Glomerulopathy.
Abstract
Immunotactoid glomerulopathy (ITG) is a rare cause of chronic kidney disease (CKD) and end-stage-renal-disease (ESRD). It is often associated with monoclonal gammopathy and/or hematologic malignancy. We report a patient originally diagnosed with ITG in 1998. He presented with nephrotic-range proteinuria, hypertension, and a gradual decline in glomerular filtration rate. A published case report of this patient at the time the disease was originally diagnosed described only a small peak of IgM paraprotein without lymphoma or plasma cell dyscrasia. He was diagnosed with monoclonal gammopathy of unknown significance. He later developed ESRD and initiated hemodialysis in 2004. Fourteen years after the diagnosis of ITG and MGUS was made he developed headache, lymphadenopathy, borderline splenomegaly, thrombocytopenia, and coagulopathy. Workup revealed a very high level of monoclonal IgM-kappa (4390 mg/dL),and low grade B-cell lymphoma, consistent with lymphoplasmacytic lymphoma, leading to a diagnosis of Waldenstrom’s macroglobulinemia (WM). He died shortly thereafter of complicated gram-negative sepsis. To our knowledge this is the first report of WM associated with ITG. The patient's course illustrates that plasma cell dyscrasia and lymphoma can present many years after the original diagnosis of ITG is made and that continued vigilance for these conditions is warranted.
Article Information
- Received
- Accepted
- Published
Academic Editor: Krzysztof Roszkowski, Nicolaus Copernicus University, PL. Department of Oncology, Radiotherapy and Gynecologic Oncology.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2015 Ezra Gabbay, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Corresponding author: Ezra Gabbay, Division of Adult Nephrology, Shaare Zedek Medical Center, Jerusalem, Israel —
Competing Interests
The authors have declared that no competing interests exist.
Funding
No specific funding statement was provided by the authors.
Data Availability
No data-availability statement was provided by the authors.
Citation:
Introduction
Immunotactoid glomerulopathy (ITG) is a well recognized, albeit very rare, cause of chronic kidney disease (CKD) and end-stage-renal disease (ESRD)1. It is often described with a similar, somewhat more common condition, fibrillary glomerulonephritis, (FGN)2, 3. Both conditions involve glomerular deposition of immunoglobulin-derived, Congo-red negative material and have similar clinical manifestations including nephrotic-range proteinuria, hematuria, hypertension, CKD and ESRD4. However, several differences between these conditions caused many investigators to consider them as separate entities. Electron microscopy (EM) in ITG shows larger (>30nm), stacked microtubules rather than smaller (16-24nm) fibrils in a random pattern in FGN. Hypocomplementemia and C3 positive deposits are common in ITG. The deposits are usually polyclonal in FGN, and monoclonal or oligoclonal in ITG3, 5, 6. This latter feature of ITG correlates with a higher incidence of paraproteinemia/monoclonal gammopathy and a stronger association with lymphoproliferative disorders compared to FGN 3, 5. We describe a patient who was initially diagnosed with ITG associated with IgM monoclonal gammopathy of unknown significance (MGUS). There was no evidence of lymphoma or plasma cell dyscrasia at the time of diagnosis. Fourteen years later, he developed clinical, biochemical and pathological features of Waldenstrom’s macrglobulinemia (WM) and lymphoplasmacytic lymphoma (LPL).
Case Report
The patient was an 83 year old Ashkenazi-Jewish man, treated with hemodialysis for 8 years. Upon transferring to our unit in 2010 he was clinically stable as an outpatient, with good functional capacity and cognitive function.
Past medical history that was known at the time of presentation to our hospital was remarkable for ITG diagnosed in 1998, presenting as new onset hypertension, pedal edema, and proteinuria of 15 grams/ day. He was treated with one course of prednisone and melphalan shortly thereafter, and then was temporarily lost to follow up. He had gradual decline in glomerular filtration rate until, in 2004, he started hemodialysis. The medical history was also remarkable for ischemic heart disease, ischemic colitis, and benign prostatic hypertrophy.
In mid 2011 he began to experience progressively worsening headaches. Cranial imaging and temporal artery duplex were negative. Two months later he was hospitalized for respiratory distress and then a fall with head trauma. Increased frequency and duration of hemodialysis treatments did not improve his symptoms. Additional findings included borderline splenomegaly, mediastinal lymphadenopathy, thrombocytopenia (with a negative test for heparin-induced thrombocytopenia), non-clotting prothrombin time and later activated partial thromboplastin time assays and hypofibrinogenemia. A month later, he was re-admitted with dyspnea, somnolence, fever and hypotension, and treated empirically with antibiotics. Blood cultures revealed gram negative bacteremia. Funduscopy showed retinal hemorrhages. Head CT demonstrated chronic subdural hematomas. A nephrology consult was requested as part of a multidisciplinary evaluation. The original diagnosis of ITG was then raised as a potential clue to his recent clinical course. A possible ITG-related hematologic disease was therefore explored. Further investigation revealed serum IgM level of 4390 mg/dL (normal 50-300), IgA level was 81.6 mg/dL (normal 70-400) and IgG 495mg/dL (normal 650-1600). Immunofixation showed monoclonal bands of IgM and kappa light chain. Free serum kappa to lambda light chain ratio was 26 (normal 0.26-1.65). C-reactive protein level was elevated (34.6 mg/dL), C3 and C4 levels were low (31 and 6 mg/dL respectively) Anti-phospholipid antibody, (predominantly IgM) was positive. Hepatitis C serology was negative.
Bone marrow (BM) biopsy showed foci of medium sized lymphocytes in an interstitial, non-nodular/non-sinusoidal pattern. These were all CD20 positive, some were CD138 positive at a medium-high intensity, all were CD5, CD10, CD23, BCL 6, cyclin D1 negative. Dutcher bodies, typical of WM, were not seen. Kappa and Lambda light chain marrow staining did not demonstrate monoclonality. A pink, homogenous, Congo-red negative material was noted in the marrow (Figure 1). CD117 positive mast cells were observed. Flow cytometry revealed a monoclonal CD19+population which constituted 17% of the total cell population and was highly positive (>90%) for CD20, CD22, CD25 and CD79b. It was moderately positive (~25%) for CD23 and FMC7 and negative for CD10, CD103 and CD5. Ten percent of this population was CD38 positive and 99% had kappa surface immunoglobulin. Based on the very high IgM level in the serum and the BM findings highly consistent with LPL, a diagnosis of WM was made.
Figure 1. Findings on paraffin – embedded bone marrow trephine biopsy sections, A- Interstitial deposits of amorphous eosinophilic Congo-red negative substance H&EX40 , B-Infiltrate of small lymphoid cells. H&E, x40, C- Cd138 immunostaining, x40, D- Cd20 immunostaining x40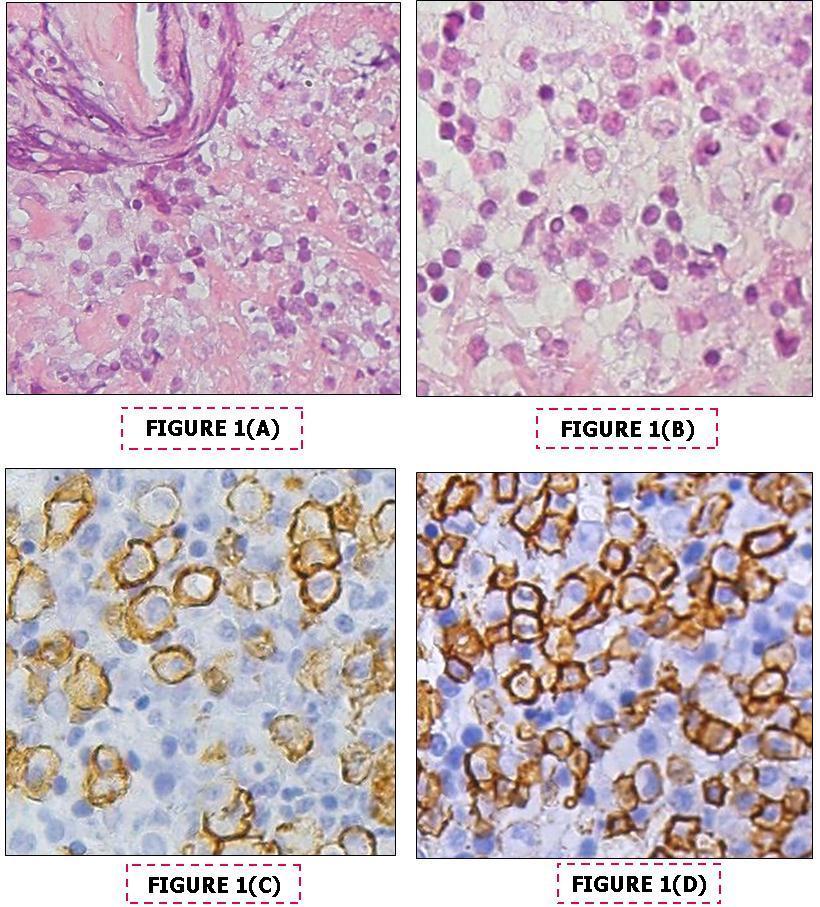
Download figure
It was then discovered that our patient had been diagnosed with IgM MGUS at the time of the initial diagnosis of ITG and that this presentation in the very same patient, along with bone marrow deposits of amyloid like material and hypocomplementemia was published as a case report in 2001 8. That report detailed the results of a kidney biopsy that showed mesangiocapillary glomerulonephritis with glomerular thrombi on light microscopy. Congo-red stain was negative. Immunofluorescence microscopy showed mesangial and lobular staining, strongly positive for IgA, C3, C4, C1q, kappa and lambda light chains; minimal for IgM and IgG. EM showed subendothelial deposits of microtubular structures, 40 nm in diameter. Additional findings detailed in the 2001 report included serum and urine electrophoresis showing a small peak in the gamma region, characterized as IgM-kappa by immunoelectrophoresis and immunofixation,and diagnosed as MGUS. Serum IgM concentration then was 275 mg/dL. Bone marrow aspirate and biopsy described in that first report revealed no evidence of lymphoproliferative disease or plasmacytic infiltration. There were Congo red-negative deposits in the BM. On EM, these deposits were similar to those found in the glomerulus8.
The patient’s clinical course in his final hospitalization was remarkable for initial improvement with antibiotics and dexamethasone, followed by another deterioration in his condition. He died approximately 2 weeks after his last admission.
Discussion
Immunotactoid glomerulopathy is more closely associated with hematologic diseases compared to FGN3, 5, 9. Recent studies did show that FGN is sometimes associated with malignancy or dysproteinemia, with multiple myeloma present in 9% of patients6, but the association with hematologic abnormalities remains much stronger in ITG. Most ITG patients exhibit either monotypic deposits, monoclonal immunoglobulin in the serum and/or lymphocytes, or lymphoproliferative disorder 7. The largest clinico-pathologic series to date (16 patients), found a serum monoclonal-spike (IgG or IgA) in 63% of ITG patients, and hematologic malignancy in 38%, including CLL in 19%, LPL13% (2 cases) and myeloma in 13% 9. In our case, the findings when the diagnosis was originally made suggested only MGUS, with no evidence of lymphoma. In the first report on this patient the authors cogently stated that the “small peak of paraprotein, …together with the hematologic parameters present led to the diagnosis of MGUS…these nephropathies may represent early renal expression of a plasma cell dyscrasia. Only clinical follow-up will indicate whether there is a correlation between the renal damage and MGUS.”8. The patient’s course illustrated that overt plasma cell dyscrasia ultimately presented itself, after an interval of 14 years. The progression of IgM MGUS to WM and other hematologic disorders has been well documented in longitudinal follow-up studies 10.
To our knowledge, this is the first report of WM associated with ITG. Kidney involvement in WM is uncommon, and usually occurs as glomerular IgM deposits, amyloidosis, cryoglobulinemia11, 12, or minimal change disease13, none of which was present when our patient was diagnosed with ITG. Mesangiocapillary glomerulonephritis, similar to the light-microscopy morphology in our patient, has been described in WM 14, but with mesangial IgM deposits. In our patient, the glomerular deposits were predominantly IgA and complement, with only minimal staining for IgM 8.
The diagnosis of WM in this patient was based on very high serum monoclonal IgM levels, and findings indicative of LPL in the BM 15. Moreover, in retrospect, plasma cell dyscrasia could have accounted for many of the chronic and subacute symptoms and signs that preceded the last hospitalization, such as headache, functional decline, lymphadenopathy, splenomegaly, thrombocytopenia and coagulopathy. While the diagnosis of marginal zone lymphoma (MZL) cannot be altogether excluded on the basis of the BM findings, it is far less likely than WM. The mean serum IgM concentration in MZL is 0.95 g/dL compared to 2 g/dL in WM, in our patient it was >4g/dL. Furthermore, the interstitial, rather than nodular/sinusoidal pattern of BM infiltration supports a diagnosis of LPL over MZL, as do the absence of CD23 staining (which is typically present in a meshwork of dendritic reticular cells in MZL), the medium to high intensity of CD138 staining and the presence of mast cells16, 17. IgM myeloma was excluded by the absence of lytic bone lesions 18.
In conclusion, the course of this patient, the first to show an association between ITG and WM, illustrates that ITG can initially present without major hematologic pathology being detected, and that overt plasma cell dyscrasia and lymphoma can become evident only much later. It underscores the importance of continued long-term follow up of ITG patients for underlying hematological malignancies. It also illustrates that investigation of clinical deterioration in a dialysis patient must consider a late manifestation of the original cause of ESRD, however many years may separate the original diagnosis and the present signs and symptoms.
References
- 1.Korbet S M, Schwartz M M.. and Lewis EJ.(1991) Immunotactoid glomerulopathy.Am J Kidney Dis.17,247-257 .
- 2.Rosenmann E.and Eliakim M.(1997) Nephrotic syndrome associated with amyloid-like glomerular. 18-301.
- 3.Rosenstock J L, Markowitz G S, Valeri A M, Sacchi G.Appel GB,et al.(2003) Fibrillaryand immunotactoid glomerulonephritis: Distinct entities with different clinical and pathologic features.Kidney. Int.63 1450-1461.
- 4.Pronovost P H, Brady H R, Gunning M E, Espinoza O.and Rennke HG.(1996) Clinical features, predictors of disease progression and results of renal transplantation in fibrillary/immunotactoid glomerulopathy. , Nephrol Dial Tranplant.11,837-842
- 5.Alpers C E, Kowalewska J. (2008) Fibrillary glomerulonephritis and immunotactoid glomerulopathy. , J Am Soc Nephrol.19 34-37.
- 6.Nasr S H, Valeri A M, Cornell L D, Sethi S, Leung N.Fibrillary glomerulonephritis: a report of 66 cases from a single institution. (2011)ClinJ AmSocNephrol.6,775-784
- 7.Bridoux F, Hugue V, Coldefy O, Goujon J M, Bauwens M.(2002)Fibrillary glomerulonephritis and immunotactoid (microtubular) glomerulopathy are associated with distinct immunologic features.Kidney Int.,621764-1775.
- 8.Da’as N, Kleinman Y, Polliack A, Amir G, Ne'eman Z.et al.(2001) Immunotactoid glomerulopathy with massive bone marrow deposits in a patient with IgM kappa monoclonal gammopathy and hypocomplementemia.Am. , J Kidney 38-335.
- 9.Nasr S H, Fidler M E, Cornell L D, Leung N.Cosio FG, et al.(2011) Immunotactoid glomerulopathy: clinicopathologic and proteomic study.NephrolDial Transplant.27,4137-4146.
- 10.Kyle R A, Benson J, Larson D, Therneau T, Dispenzieri A. (2009) . IgM Monoclonal Gammopathy of Undetermined Significance and Smoldering Waldenström’s Macroglobulinemia. Clin Lymphoma Myeloma9 17-18.
- 11.Morel-Maroger L, Basch A, Danon F, Verroust P.. and Richet G.(1970) Pathology of the kidney in Waldenström's macroglobulinemia. Study of sixteen 283-123.
- 13.Hory B, Saunier F, Wolff R, Saint-Hillier Y, Coulon G.et al.(1987) Waldenstrom macroglobulinemia and nephritic syndrome with minimal change. 45-68.
- 14.Veltman G A, S van Veen, Kluin-Nelemans J C, Bruijn J A, van.. Es LA.(1997) Renal disease in Waldenstrom’s macroglobulinaemia.NephrolDial Transplant 12, 1256-1259.
- 15.Owen R G, Treon S P, Al-Katib A, Fonseca R, Greipp P R. (2003) . Clinicopathological definition of Waldenstrom's macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom's Macroglobulinemia.SeminOncol 30-110.
- 16.Arcaini L, Varettoni M, Boveri E, Orlandi E, Rattotti S.et al.(2011) Distinctive clinical and histological features of Waldenström's macroglobulinemia and splenic marginal zone lymphoma.ClinLymphoma MyelomaLeuk.11,103-105.
Cited by (2)
This article has been cited by 2 scholarly works according to:
Citing Articles:
Wolfgang Neukirchen, A. Oesterling, D. Wennmann, B. Heitplatz, Peter Ritter et al. - Case Reports in Nephrology and Dialysis (2022) Semantic Scholar
Clinical Case Reports (2020) OpenAlex Crossref Semantic Scholar