Laboratory Tests Used in the Diagnosis of Immune Thrombocytopenia and General Treatment Approaches
Abstract
Immune thrombocytopenia currently called under its’ new name, immune thrombocytopenic purpura (ITP) is a disease characterized by thrombocytopenia, in which the body attacks its own platelets due to the disorders in immune system. The pathophysiology of this disease includes increased platelet destruction and most megakaryocyte production in bone marrow. The most common clinical manifestation of ITP is mild or severe progressive bleeding that could result in death. ITP is generally named as primary or secondary ITP according to thrombocytopenia severity, disease duration, bleeding status and secondary occurrence of the disease. Currently for diagnosis, despite the blood count, antiglobulin test and laboratory tests that can detect platelet-bound antibodies, they are not enough for definitive diagnosis. Like the difficulty in diagnosis, ITP treatment is quite complicated which varies depending on age, characteristics and risk of the patient. It is classified as first, second and third-line treatment options. Also, depending on the condition of patients, combined treatment might be an option which increases the complexity of the treatment. Unfortunately, discussions related to different clinical applications in diagnosis and treatments continue recently. For this reason, we considered that preparation of a review containing recent updates in diagnostic approaches and treatment options in ITP will be remarkable and beneficial for physicians interested in this subject.
Author Contributions
Academic Editor: Rada M. Grubovic, Head of Department for Stem Cell Collection President of Macedonian Society for Transfusion Medicine, Macedonia.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2020 Fatih Ozcelik, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Platelets are the smallest shaped elements of the blood that play a primary role in preventing bleeding. One of the most common diseases associated with platelets is immune thrombocytopenia (ITP), an autoimmune disease, formerly called immune thrombocytopenic purpura. ITP is a disease in which autoantibodies attack the body's own platelets. These auto-antibodies, which are formed against antigens on the platelet surface, are formed as a result of a person's own tolerance disorder against their own platelet antigens due to disorders in the immune system. Although the pathophysiology of ITP is not yet fully understood, most important point is considered to be the production of antiplatelet auto-antibodies. Normally our immune system recognizes foreign organisms (bacteria, viruses, cancer cells and other foreign substances) and releases substances called antibodies against them. These antibodies form complexes by marking foreign organisms. Monocytes / macrophages recognize these complexes and destroy these marked complexes. In ITP, the immune system marks the surface antigens of platelets as foreign for an unknown reason. This situation leads to opsonization and disintegration of platelets in the organs (reticuloendothelial system) that form the body's defense mechanism, especially the spleen 1,2,3.
The incidence of ITP in adults is approximately 3-4 per 100.000 4,5. There are two important mechanisms in pathophysiology of the ITP. One is increased platelet destruction and the other is inadequate megakaryocyte production in bone marrow. The cause of platelet destruction is the formation of auto-antibodies targeting glycoprotein IIb/IIIa on the surface of the platelets, which occurs when immune system loses its tolerance to its platelet antigens. Marking platelets with these auto-antibodies causes them to be destroyed by macrophages or cytotoxic T cells. The decrease in megakaryocyte production is due to the dysfunction of megakaryocytes observed in ITP and insufficient levels of thrombopoietin (TPO), the main regulator of platelet production in megakoryocytes. Thus, an increase in platelet destruction and a decrease in platelet production are added 6, 7, 8. The common clinical manifestation of this condition is bleeding. The most serious bleeding is intracranial hemorrhage, which can cause stroke and death.
General Approaches and Classification in ITP
Because ITP is developing due to a plasma protein (Ig or complement), some researchers administered ITP plasma infusion to healthy individuals in a study. After the procedure, they observed a sudden drop in platelet count and this finding was considered as an evidence of existence of auto-antibodies against platelets in ITP 9.
ITP might develop secondary to autoimmune diseases such as systemic lupus erythematosus and Hashimoto's thyroiditis, as well as due to some condition such as some cancers, immune system disorders, infections (hepatitis C virus (HCV), hepatitis B virus (HBV), human immunodeficiency virus (HIV), parvovirus, cytomegalovirus (CMV), helicobacter pylori infection, tuberculosis and brucellosis), vaccines, some medications, and pregnancy (Table 1). In these situations, the disease is called secondary ITP. As a matter of course, secondary ITP treatment mainly focused on resolving underlying causes, reducing platelet destruction and stimulating platelet production. If immune thrombocytopenia occurs alone, it is called primary immune thrombocytopenia or ITP. Primary ITP is a diagnosis of exclusion and accounts for 80% of all cases. ITP was previously used as an acronym for ‘idiopathic thrombocytopenic purpura’. However, in 2009, Rodeghiero et al. prepared an international consensus report where they changed ITP as an abbreviation of immune thrombocytopenia on the grounds that most patients did not have purpura 2, 10, 11, 12, 13, 14, 15. It was also emphasized that the diagnosis of primary ITP should be diagnosed in the first 3 months of the disease. The disease is called persistent ITP if it was diagnosed within 3-12 months from the onset of the disease, and chronic ITP after 12 months. Similarly, Provan et al. followed this nomenclature in their report on the investigation of primary ITP 16.
Table 1. Secondary ITP causes| Autoimmune Disorders | Infections | Vaccinations | Drugs and pregnancy |
|---|---|---|---|
| SLE | HIV | Measles | Quinine |
| APS | HCV | Mumps | Heparin |
| Hashimoto’s thyroiditis | HBV | Rubella | Fiban |
| Grave's disease | CMV | Varicella | Abciximab |
| Rheumatoid arthritis | Rubella | Hepatitis A | Beta-lactam |
| Evans syndrome | EBV | Diphtheria | antibiotics |
| Parvovirus | Tetanus | Ristocetin | |
| H. pylori | Pertussis | Ticlopidine | |
| Clopidogrel |
The course of ITP observed in children and adults is different and its treatment is also different. ITP is mostly chronic and more severe in adults. On the contrary, it is not usually chronic in children, it occurs acutely after an infection or vaccination and usually heals spontaneously. The ITP, which is seen as a child, generally heals within the first year. However, in some children the situation is different. There is no improvement and ITP becomes chronic. Treatment options should be reconsidered in these pediatric patients. Because, in addition to the side effects (weight gain, swelling of the face, flushing, fat accumulation in the trunk, thinning of the arms and legs, thinning of the skin, flushing, purple cracks in the abdomen, bone head aseptic necrosis, hypertension, tendency to fungal infections) associated with steroid use in children, growth retardation or slowing is added. For this reason, it should be tried not to use high dose for a long time especially in chronic ITP patients. In adults, ITP has a sneaky onset and the age of its appearance ranges from 30-60 years. Its incidence is almost equal in men and women 17, 18, 19. In 2019, a document was published in which many researches and recommendations for ITP treatment were examined and evaluated. This final document provides recommendations on the diagnosis and management of ITP and quality of life assessments in adults, pregnancy and children 19.
Laboratory Methods Used in ITP Diagnosis
ITP is mostly detected incidentally by a hemogram test or bleeding story and low platelet count (thrombocytopenia) is the main finding. Thrombocytopenia may cause bleeding in mucous membranes (in mouth, lips, nasal mucosa and/or gingiva) and hematuria, melena, hypermenorrhea, petechia under the skin and rash in red-purple color following mild trauma. Although the normal platelet count range is considered as 150.000-450.000/mm3, the platelet count should be lower than 100.000/mm3 in order to descirbe it as thrombocytopenia. Because without falling below this number, there is generally no impairment in platelet functions and a bleeding diathesis. Excluding other causes of thrombocytopenia, this threshold is also used for the diagnosis of ITP. Spontaneous bleeding frequently observed in those with a platelet count lower than 30,000 / mm3. The presence of coexisting diseases (such as uremia and cirrhosis), use of some drugs (such as aspirin, heparin and warfarin), trauma, tooth extraction and surgical procedures increase the risk of bleeding 15, 16, 17.
In the case of thrombocytopenia, aggregates of the platelets could hardly form a functional plug. Accordingly, bleeding, especially after physical trauma, could take a long time and might be life threatening. Not every thrombocytopenia should be defined as ITP. Therefore, other diseases that may cause thrombocytopenia should be excluded before final diagnosis. In other words, patients should not have any complaint or finding other than bleeding during the diagnosic approaches. Primary ITP should not be a concern in the presence of symptoms such as fever, weight loss, sweating, abdominal distention, joint pain, skeletal abnormalities, mouth sores and jaundice. In the anamnesis and history, infectious and autoimmune diseases, viral and bacterial hepatitis and pregnancy that may cause secondary immune thrombocytopenia should be investigated. In addition, hereditary thrombocytopenia should be questioned while taking family history. Because when secondary causes disappear, platelet count will generally return to normal levels. Therefore, treatment strategy should be adjusted properly 15, 16, 20, 21, 22.
Some biochemical tests, viral infection screening, bone marrow examination and abdominal ultrasonography may be required to confirm the diagnosis. In addition, there are specific tests today that detect anti-platelet autoantibodies that can be used in the diagnosis of ITP, as in autoimmune hemolytic anemia (AIHA). However, there is no ideal test for ITP yet 23. Although there are many similarities between AIHA and ITP, anti-erythrocyte antibodies can be detected in the majority of AIHA patients with some simple methods (direct or indirect antiglobulin tests) 24,25, whereas platelet autoantibodies are much more difficult to detect. For this reason, there may be a contradiction in many ITP patients and definitive diagnosis can be quite challenging. As a general approach, severe thrombocytopenic response to intravenous immunoglobulin administiration is a definitive indication of ITP. However, in many patients, the diagnosis may remain uncertain. Therefore, patients with a preliminary diagnosis of ITP should be investigated for underlying secondary causes of ITP and other causes of thrombocytopenia. Table 2 summarizes the strategy of approach in this regard.
Table 2. Diagnostic approach in patients suspected of primer/seconder ITP| In the first step, evaluation of symptoms, history and physical examination | In the second step, appropriate laboratory tests | In the third step, response to therapy |
| Easy or excessive bruising | CBC, Reticulocyte count | IVIg |
| Petechiae, purpura | Peripheral blood smear | Oral steroid |
| Bleeding in the gum or nasal mucosa | HIV, HCV, HBV | Rituximab and thrombopoietin receptor antagonists |
| Blood in urine | Quantitative immunoglobulins | *Splenectomy |
| Unusually heavy menstrual bleeding | DAT | |
| Lymphadenopathy, and palpable liver or spleen | In adults, H. pylori tests (C14 urea breath test) | |
| Are there other causes of thrombocytopenia? | In age ≥60 years or haematological disorders, bone marrow test | |
| MPV, electrolytes, liver and kidney tests, protein electrophoresis | ||
| Autoantibody tests related to autoimmune diseases (ANA, ANCA, Anti Tg etc.) | ||
| Further biochemical tests based on history and abdominal ultrasound (for splenomegaly etc.) |
Reticulated platelets and immature platelet fraction interpreted in favor of ITP can be detected with flow cytometry and automated blood count device. However, to a lesser extent, reticulated platelets and immature platelets can be detected in other thrombocytopenic diseases 26. Coombs tests, which are widely used in the diagnosis of AIHA and Evans syndrome (coexistence of autoimmune hemolytic anemia and ITP), can also be applied in platelet antibodies. However, platelets cannot be isolated as easily as erythrocytes, they cannot be washed, and their numbers are relatively low. Therefore, investigation of platelets is much more difficult compared to erythrocytes. Despite all these disadvantages, Coombs tests used in the research of anti-platelet antibodies made important contributions to the understanding of ITP pathogenesis 9. In direct methods used for this purpose, immunoglobulins (or complements) bound to the platelet surface are detected, just like in erythrocytes (Figure 1). In addition, radioactive labeled Coombs antiglobulin test, which can be used in the diagnosis and follow-up of immune thrombocytopenia in adults and children, can easily detect IgG and C3 bound to platelets 27. The indirect method requires one more step than the direct method. Anti-platelet antibodies (or complement) in patients’ serum are first incubated with platelet or platelet glycoproteins for a certain period of time, and then bound with Coombs antibodies 28. In this method, trace amounts of thrombin can cause false positives because thrombin can activate platelets and cause their clustering. Therefore, thrombin must be removed with additional procedures.
Figure 1.Detection of platelet antibodies by Coombs test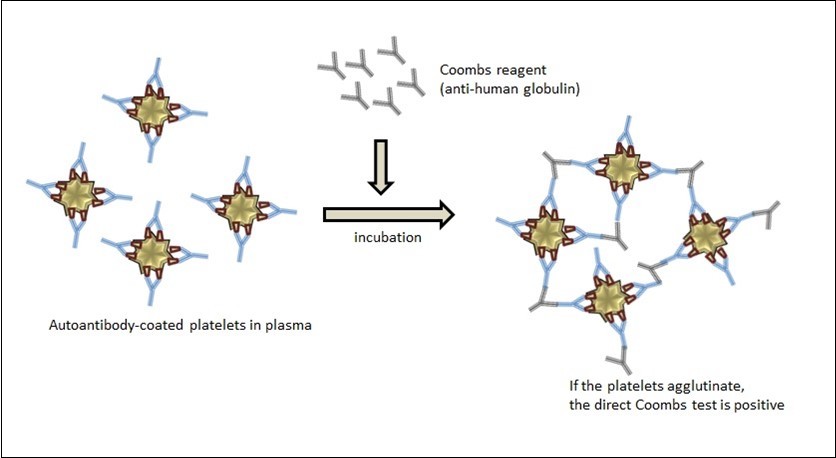
Today, different methods have been developed in which IgGs related to platelets are directly measured. In these methods, direct binding of labeled antibodies bound to platelet surface is detected by radioisotopes or flow cytometry. However, it should be remembered that most of the IgGs on the platelet surface are not pathological, and there are many IgGs in the alpha-granules in the platelet 29,30. In addition, the methods that directly measure thrombocyte-bound IgGs showed similar positive results in almost all immune and non-immune thrombocytopenic disorders, which caused these methods to be questioned and overlooked. Today, there are a limited number of laboratories where these methods are used. Monoclonal antibody-specific immobilization of platelet antigens and enzyme-linked immunosorbent assay (ELISA) methods are the most common preferred methods to detect GP IIb/IIIa (where vWF and fibrinogen bind) and/or GP Ib/IX (where vWF connects) -specific autoantibodies on the platelet surface in patients with ITP 23,31. Many laboratories researching ITP use these methods. However, these serological methods were found to have a high specificity and low sensitivity. In other words, although the positive determination of these tests can be interpreted in favor of ITP, they cannot be used as an exclusion criterion for ITP. However, there is evidence that the presence of antibodies against platelet glycoproteins can be used to predict the response to treatment 32, 33, 34.
Treatment Options of ITP
It is completely known that the immune system reactions in ITP is a pathological process in which autoantibodies are formed against platelet antigens due to impaired tolerance of their cell antigens. Based on this information, steroid, intravenous immune globulin (IVIg) and rituximab are used as important options in the treatment in order to silence or slow the immune reaction 23, 35, 36, 37, 38. Fostamatinib, which has recently been approved, has started to be among the treatment options. However, all these treatments make a temporary improvement in patients. There is no definitive treatment; thus relapses (recurrences) are frequently observed. Among alternative treatments, splenectomy is considered as an important therapeutic alternative. Ineffective results from these approaches forced scientists to find different options. While discussing which treatment method is more successful, we also find it useful to provide ideas that can shed light on studies. The scientific world has not yet found a definitive answer to why the immune system loses the tolerance to its’ tissue antigens. Therefore, more comprehensive investigations should be focused on this point. In our opinion, the most effective solution for all autoimmune diseases is to eliminate the source that will lead to the disorder.
Due to the high risk of morbidity and mortality, it is recommended to start ITP treatment in adults whose platelet count below 30,000 / mm3 and in patients with severe bleeding, regardless of platelet count. ITP's first-line treatment options include corticosteroids, IVIg, and anti-D immune globulin (in patients with Rh positive blood group) treatment. These options can quickly restore platelet count in emergency situations. However, these treatment options are not suitable for long-term treatment because of their limited response times and long-term toxicity. Their use also requires strict monitoring. Usually corticosteroid therapy is the first choice for the initial treatment of ITP due to its easy-to-apply and low cost 15, 39, 40. However, IVIg therapy, which can increase the number of platelets more rapidly, should be preferred in patients with active bleeding. Increased platelet count is expected within the first 24-48 hours of corticosteroid treatment.
Corticosteroids
The main purpose of the use of corticosteroids in ITP is to reduce antibody production by immunosuppression and prevent platelet destruction by macrophages / monocytes. However, it should be remembered that these drugs have serious side effects. For this purpose, prednisone, prednisolone, methylprednisolone and dexamethasone, which are medium and long-acting steroids, are used. In the treatment of ITP, prednisone, prednisolone, methylprednisolone and dexamethasone are used 40,41. The most common approach is the use of oral prednisone at a dose of 0.5-2.0 mg / kg for 2-4 weeks, followed by 40 mg / day dexamethasone 4 days a week for 2 - 4 weeks with 1-4 cycles. In a study comparing the efficacy and reliability of treatment with high-dose dexamethasone and prednisone in patients newly diagnosed ITP, a group of patients was administered 40 mg dexamethasone per day for 4 days or 1 mg / kg prednisone per day for four weeks. The initial response of dexamethasone (reaching the platelet count greater than 30,000 / mm3) was faster than prednisone. However, there was no difference between them in terms of continuous response (reaching a platelet count greater than 30,000 / mm3 for six months) 41. Unfortunately, long-term corticosteroid therapy has several important side effects such as osteoporosis, diabetes, hypertension and weight gain. For this reason, they should be avoided as much as possible and the patients should be followed closely if their use was mandatory. If there is not an enough response with corticosteroids, IVIg is recommended as a second-line therapy. The same approach can be applied in patients who cannot tolerate the side effects of corticosteroids 42.
Intravenous Immunoglobulin
The main purpose in the use of IVIg therapy is to shut down the Fc receptors in the reticuloendothelial system, where platelet destruction occurs, to prevent the destruction of labeled platelets by phagocytes. The standard IVIG dose has been reported to be 400 mg/kg/day for 5 days. The most guidelines currently recommend administering a single dose of IVIg (1 g/kg), which can be repeated depending on the platelet response. However, IVIg treatment is temporary and some side effects may occur; IVIg administration is generally well tolerated. Headaches, chills, arthralgia, back pain and rarely occurring kidney injury are the most common side effects 13, 20, 43, 44. The risk of kidney injury can be reduced by adequate hydration before IVIg administration.
Anti-D Immunglobulin
Anti-D immunoglobulin therapy has been used for the treatment of immune thrombocytopenia in non-splenectomized Rh-positive patients and this treatment was approved by the Food and Drug Administration (FDA). IVIg prepared from the plasma of immunized Rh-negative human donors can be used as an alternative for patients with Rh-positive blood type. The goal of this treatment is to neutralize the binding of autoantibodies to platelets and block the macrophage system. The purpose of this treatment is to block the macrophage system by neutralizing the binding of autoantibodies to platelets. The recommended dose for anti-D immune globulin ranges from 50-75 μg / kg intravenously at one time. The side effects of anti-D immune globulin therapy are similar to those of IVIg. Fatal inravascular hemolysis cases have also been reported. Because of these serious side effects, patients receiving anti-D immune globulin should be monitored for at least eight hours for signs of inravascular hemolysis and other side effects 45, 46, 47.
Splenectomy
Second and third-line treatments should be applied to patients with permanent and chronic ITP who have a high risk of bleeding that does not require emergency or rescue treatment. While rituximab is recommended as the second-line treatment at this stage, splenectomy is recommended in the third-line treatment. While splenectomy was among the second-line treatment options, it was replaced in the third-line treatment due to the effective novel treatments that emerged with TPO receptor agonists (eltromobopag and rhomiplasty) 48, 49, 50. Therefore, splenectomy is considered an option following unsuccessful steroid and rituximab treatment. In addition, the response rate of the patients to splenectomy (especially higher in young patients) is 2/3, which is actually the answer to why it should be in third-line 40, 51, 52. In cases where a certain response cannot be obtained with monotherapies, second and third-line treatments could also be considered in combination. Splenectomy decision should be considered well because it is necessary to allow spontaneous or treatment-related remissions to occur. Therefore, splenectomy should be postponed until 12 months after the diagnosis of ITP, especially in children and elderly patients with high surgical morbidity 53,54.
In general, splenectomy is considered as an effective therapeutic option in ITP, but it has significant complications. One of the most important complications of splenectomy is bleeding during the procedure. Other complications include venous thromboembolism, pneumonia, and other infections. Splenectomy-related mortality rate is lower in laporoscopy compared to open laparatomy (1% and 0.2%, respectively) 51, 55, 56. In addition, relapses can be observed after splenectomy. Therefore, radiographic evaluation should be performed for the possibility of accessory spleen. As a hope, immunosuppressive treatment trials are among the options in cases where there is no response to splenectomy. These immunosuppressive agents include cyclophosphamide, azathioprine, cyclosporine and vincristine. In later stages, use of plasmapheresis, androgens (danazol), colchicine and dapson may be on the agenda 57, 58, 59.
Thrombopoietin Receptor Agonists
It has been reported that TPO-receptor agonists (eltrombopag, romiplostim and avatrombopag), a class of platelet growth factors, can be used in patients with chronic ITP who responded poorly to corticosteroids, immunoglobulins, and splenectomy. TPO-receptor agonists induce platelet production by activating JAK2 and STAT5 kinase pathways through TPO receptors on megakaryocytes. Megakaryocytes induced in this way will have longer lives and produce more platelets. Thus, they reverse the production defect that creates a ground for ITP 1, 13, 60, 61, 62, 63. Besides immunosuppressive, immunomodulating agents and splenectomy, TPO-receptor agonists is a new option and limited to patients with chronic ITP who failed with glucocorticoids, splenectomy and IVIg 15,63. However, the use of these agents is common in patients with acute or persistent ITP, and the use as a first-line is still under investigation 64. In order to provide optimal benefit with TPO-receptor agonists in treatment-resistant ITP patients, it is necessary to know how to use, dosage, duration and side effects in all treatment steps and combination therapy because the decision to administer a TPO-receptor agonist after an immunosuppressive therapy and splenectomy may be complicated. In general, TPO-receptor agonists that require long-term use have a higher clinical response rate (~ 80%) than second and third-line treatment agents 65. It is commonly observed that ITP patients with lower TPO levels better respond to TPO-receptor agonists 66,67. It is also thought that TPO levels can be used to guide the treatment. It was found remarkable that in patients who have not used TPO-receptor agonists and had splenectomy, a response rate up to 50% has been observed, and a limited number of patients developed intolerance (tachyphylaxis) to the commercial dose of TPO-receptor agonists 68, 69, 70. In second-line therapy, rituximab and then splenectomy are seen as an alternative to TPO-receptor agonists 71. However, in many clinical applications, it is considerable to use medical treatments for at least 1 year before considering splenectomy in most adults.
Romiplostim is a subcutaneous peptide that administered once a week. In order to get sufficient response to Romiplostim (platelet number is ≥50.000 / mm3) 1-2 µg / kg / week starting dose is sufficient and treatment is continued with 1µg / kg every week 72. It is reported that in patients with acute bleeding symptoms and refractory disease despite glucocorticoid and IVIg, romiplostime can be started at higher doses in order to obtain more prudent and effective results in a shorter time 73. With the classical approach, the gradual increase in the dose of romiplostim may cause deep thrombocytopenia and bleeding risk in a long time, while a faster dose increase may result in thrombocytosis (risk of thromboembolism).
Eltrombopag and avatrombopag are small molecule TPO-receptor agonists that can be administered orally once a day. The recommended starting oral dose of Eltrombopag is 50 mg per day under fasting. The initial dose of eltrombopag is 25 mg in patients with chronic liver disease and in children aged 1-5 years 62,63,74. It is used in higher doses (150 mg/day) in aplastic anemia. However, there is not reliable data to support its use at this level in patients with ITP. In alternate intermittent dosing, the dose of 75 mg is usually used 1-5 times a week instead of the daily dose, due to the long half-life of the eltrombopag (26-36 hours). In patients receiving romiplostim or eltrombopag, it is recommended to reduce the drug dose when the platelet counts reach a level greater than 400,000 / mm3. Eltrombopag is potentially hepatotoxic 75. Therefore, it should not be used in patients with ITP and chronic liver disease, as it will increase the risk of venous thromboembolism (VTE).
Avatrombopag is not yet approved for use in ITP treatment. Therefore, there are no general dosage recommendations. However, in phase II studies, it is reported that a daily dose of 5-10 mg/day creates an adequate platelet response in approximately ½ of patients, and a dose of 20 mg/day in 80% of patients 76.
Rituximab
CD19, CD20 and CD22 are antigens that are not expressed in non-lymphoid cells and expressed on the surface of all B cells CD19 antigen is also expressed by follicular dendritic cells. Therefore, the CD20 antigen is considered an antibody dependent antitumor target. The rituximab, a monoclonal anti-CD20 antibody produced for this purpose, is commonly used in non-Hodgkin lymphomas. The basic approach in Rituximab therapy is to prevent B cells from producing autoantibodies. FDA approved its use in the treatment of non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, and rheumatoid arthritis. However, since the studies are not finished, there is no approval yet for the use of rituximab in ITP. It is recommended that IVIg can be used in patients with high risk of bleeding that has failed with the treatment of anti-D immune globulin and corticosteroids. The recommended dose of rituximab in ITP is 375 mg/m2 once a week for four weeks 1, 40, 77, 78, 79, 80. In adults with chronic disease and ITP, the platelet response of rituximab has been reported to be approximately 60%, especially in women and patients under 40 years of age. Its use was found to be limited in emergency and acute conditions. It has also been observed that rituximab is associated with some side effects such as fever, allergic skin rashes, tremors, and vomiting.
Fostamatinib
Fostamatinib is an oral inhibitory agent (Moore) of spleen tyrosine kinase (Syk), which is expressed by hematopoietic cells and plays an important role in the destruction of platelets by Fc-receptor activation 81. Syk signaling pathway is the center of phagocytosis-based antibody-mediated platelet destruction in adults with ITP. In two different studies conducted in 2018 and 2019, it was found that approximately 30% of patients using phosphatimib received a platelet response (platelet count ≥ 50 000 / μL) within the first 12 weeks of treatment. Although generally well tolerated, some non-permanent side effects of this treatment have been observed, such as diarrhea, hypertension, nausea, dizziness and increased liver function tests. In this multicenter study, it was found that the treatment of fostamatinib showed significant success even in those who failed splenectomy, thrombopoietin agonist and rituximab treatment 82,83. Co-administration of phosphomatinib, which is mainly metabolized via the Cytochrome P450 3A4 pathway in the liver and intestine, with Cytochrome P450 3A4 inhibitors or inducers, which use the same metabolic pathway, can change the effects of fostamatinib 81. Therefore, it should be used carefully.
Fostamatinib has been approved in the U.S. for use in adult patients with chronic ITP who have responded poorly to a previous treatment 84. However, there is still insufficient experience in its use. Therefore, we think that it will be useful to continue the research.
Criteria for Confirming ITP Diagnosis and Treatment
In the light of all the abovementioned evaluations and literature reviews, we prefer to say that there is still no single laboratory test that can be alone for the definitive diagnosis of ITP. Therefore, the diagnosis should be made with both clinical and laboratory evaluations. Attention should be paid to pseudothrombocytopenias, which may lead to an incorrect diagnosis of ITP. In order to discriminate the true pseudothrombocytopenia from thrombocytopenia, the use of other anticoagulated (sodium citrate, oxalate and heparin) tubes instead of EDTA tubes, bringing the blood to 37 °C, determining the platelet aggregation under the microscope or using the kanamycin added blood samples 85,86. The presence of thrombocytopenia, exclusion of other causes, detection of anti-platelet autoantibodies and increase in the number of platelets in response to treatment (steroid, IVIg, and secondary causes) should be the criteria to be used for definitive diagnosis 23, 35, 36, 37. However, a relatively lower sensitivity of the methods for the detection of anti-platelet autoantibodies and their inability to exclude ITP make other two suggestions more valuable in the diagnosis.
Steroids remain important as a first-line therapeutic option in clinical practice. Rituximab and TPO-receptor agonists with limited side effects, which are alternatives to IVIg, anti-D immune globulin and splenectomy, are highly effective therapeutic option in the management of ITP. When choosing the first- (corticosteroids, IVIG and anti-D immune globulin), second- or third-line treatment options mentioned above, the patients’ bleeding risk should be considered, and an individual treatment algorithm should be planned. Treatment of patients with mild ITP might begin with corticosteroids as a general approach. IVIg therapy (as an alternative, anti-D immune globulin) should be reserved for the patients with severe bleeding and / or those who do not develop enough response to corticosteroids. In patients with treatment-resistant and non-urgent chronic ITP, splenectomy and rituximab are the recommended options. Furthermore, TPO-receptor agonists can also be considered as an additional therapy in the treatment of chronic ITP. Beyond all these therapeutic options, combined treatment should not be also ignored.
Conclusion
In conclusion, both the clinical and laboratory evaluation seems to be the only solution in the diagnosis of ITP. Thrombocytopenia and platelet response to treatment continues to be the most effective diagnostic criteria. There is no specific treatment pattern for ITP. The treatment in ITP continues to be a complicated that uses different options from monotherapy to combined therapy and adjusted according to the patients’ conditions.
Abbreviations
SLE: Systemic lupus erythematosus
APS: Antiphospholipid syndrome
HIV: Human immunodeficiency virus
HCV: Hepatitis C virus
HBV: Hepatitis B virus
CMV: Cytomegalovirus
EBV: Epstein-Barr virus
H. Pylori: Helicobacter pylori
ITP: Immune thrombocytopenia
IVIg: Intravenous immunoglobulin
CBC: Complete blood count
HBV: Hepatitis B virus
HCV: Hepatitis C virus
Human immunodeficiency virus
DAT: Direct antiglobulin test
MPV: Mean platelet volume
ANCA: Antineutrophil cytoplasmic antibody
ANA: Antinuclear antibody
Tg: Tyroglobulin
References
- 1.Zufferey A, Kapur R, J P Semple. (2017) athogenesis and Therapeutic Mechanisms in Immune Thrombocytopenia (ITP). , J Clin Med 6(2), 16.
- 2.McMillan R. (2017) The Pathogenesis of Chronic Immune Thrombocytopenic Purpura. Semin Hematol.44(SUPPL5):. 3-11.
- 3.George J N, Woolf S H, Raskob G E. (1996) Idiopathic thrombocytopenic purpura: A practice guideline developed by explicit methods for the American Society of Hematology. , Blood 88(1), 3-40.
- 4.Lambert M P, Gernsheimer T B. (2017) Clinical updates in adult immune thrombocytopenia. , Blood 129(21), 2829-2835.
- 5.Abadi U, Yarchovsky-Dolberg O, Ellis M H. (2015) Immune Thrombocytopenia. , Clin Appl Thromb 21(5), 397-404.
- 6.Nugent D, McMillan R, Nichol J L, Slichter S J. (2009) Pathogenesis of chronic immune thrombocytopenia: increased platelet destruction and/or decreased platelet production. , Br J Haematol 146(6), 585-596.
- 7.Godeau B. (2014) Immune thrombocytopenic purpura: Major progress in knowledge of the pathophysiology and the therapeutic strategy, but still a lot of issues. , Presse Med 43(4), 47-48.
- 8.Tripathi A K, Mishra S, Kumar A, Yadav D, Shukla A et al. (2014) Megakaryocyte morphology and its impact in predicting response to steroid in immune thrombocytopenia. , Platelets 25(7), 526-531.
- 9.Harrington W J, Minnich V, J W Hollingsworth, C V Moore. (1990) Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. http://www.ncbi.nlm.nih.gov/pubmed/2187937. AccessedFebruary12,2020 , J Lab Clin Med 115(5), 636-645.
- 10.Tahir H, Sheraz F, Sagi J, Daruwalla V. (2016) Immune Thrombocytopenia (ITP) Secondary to Subclinical Hashimoto’s Thyroiditis. J Investig Med High Impact Case Reports. 4(2), 232470961664708.
- 11.Reese J A, Li X, Hauben M. (2010) Identifying drugs that cause acute thrombocytopenia: an analysis using 3 distinct methods. , Blood 116(12), 2127-2133.
- 12.Cecinati V, Principi N, Brescia L, Giordano P, Esposito S. (2013) Vaccine administration and the development of immune thrombocytopenic purpura in children. Hum Vaccin Immunother 9(5), 1158-1162.
- 13.Imbach P, Crowther M. (2011) Thrombopoietin-receptor agonists for primary immune thrombocytopenia. , N Engl J Med 365(8), 734-741.
- 14.Malfertheiner P, Megraud F, O’Morain C A. (2017) . Management of Helicobacter pylori infection-the Maastricht V/Florence Consensus Report. Gut 66(1), 6-30.
- 15.Rodeghiero F, Stasi R, Gernsheimer T. (2009) Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. , Blood 113(11), 2386-2393.
- 16.Provan D, Stasi R, Newland A C. (2010) International consensus report on the investigation and management of primary immune thrombocytopenia. , Blood 115(2), 168-186.
- 17.Diz-Küçükkaya R, Chen J, Geddis A, López J A. (2010) . Williams Hematology. Eighth. USA: McGraw-Hill Companies, Inc. https://accessmedicine.mhmedical.com/content.aspx?bookid=358§ionid=39835943. Accessed February 12 Thrombocytopenia. Içinde: Lichtman, Marshall A.; Kipps, Thomas J.; Seligsohn, Uri; Kaushansky, Kenneth; Prchal JT, ed .
- 18.McCrae K. (2011) Immune thrombocytopenia: No longer “idiopathic”. , Cleve Clin J Med 78(6), 358-373.
- 19.Provan D, Arnold D M, Bussel J B. (2019) Updated international consensus report on the investigation and management of primary immune thrombocytopenia. , Blood Adv 3(22), 3780-3817.
- 20.Neunert C, Lim W, Crowther M, Cohen A, Solberg L et al. (2011) The American Society of Hematology. , Blood 117(16), 4190-4207.
- 21. (2003) British Committee for Standards in Haematology General Haematology Task Force. Guidelines for the investigation and management of idiopathic thrombocytopenic purpura in adults, children and in pregnancy. , Br J Haematol 120(4), 574-596.
- 22.Arnold D M, Bernotas A, Nazi I. (2009) Platelet count response to H. pylori treatment in patients with immune thrombocytopenic purpura with and without H. pylori infection: a systematic review. , Haematologica 94(6), 850-856.
- 23.Kelton J G, Vrbensky J R, Arnold D M. (2018) How do we diagnose immune thrombocytopenia in 2018?. , Hematology 2018(1), 561-567.
- 24.Zantek N D, Koepsell S A, Tharp D R, Cohn C S. (2012) The direct antiglobulin test: A critical step in the evaluation of hemolysis. , Am J Hematol 87(7), 707-709.
- 26.Pons I, Monteagudo M, Lucchetti G. (2010) Correlation between immature platelet fraction and reticulated platelets. Usefulness in the etiology diagnosis of thrombocytopenia. , Eur J Haematol 85(2), 158-163.
- 28.Warner M, Kelton J G. (1997) Laboratory investigation of immune thrombocytopenia. , J Clin Pathol 50(1), 5-12.
- 29.George J N, Saucerman S. (1988) Platelet IgG, IgA, IgM, and albumin: correlation of platelet and plasma concentrations in normal subjects and in patients with ITP or dysproteinemia. http://www.ncbi.nlm.nih.gov/pubmed/3390611. AccessedFebruary11,2020 , Blood 72(1), 362-365.
- 30.Kelton J G, Powers P J, Carter C J. (1982) A prospective study of the usefulness of the measurement of platelet-associated IgG for the diagnosis of idiopathic thrombocytopenic purpura. http://www.ncbi.nlm.nih.gov/pubmed/6889450. AccessedFebruary11,2020 , Blood 60(4), 1050-1053.
- 31.Naimushin Y A, A V Mazurov. (2004) Von Willebrand factor can support platelet aggregation via interaction with activated GPIIb-IIIa and GPIb. , Platelets 15(7), 419-425.
- 32.Zeng Q, Zhu L, Tao L. (2012) Relative efficacy of steroid therapy in immune thrombocytopenia mediated by anti-platelet GPIIbIIIa versus GPIbα antibodies. , Am J Hematol 87(2), 206-208.
- 33.Peng J, Ma S-H, Liu J. (2014) Association of autoantibody specificity and response to intravenous immunoglobulin G therapy in immune thrombocytopenia: a multicenter cohort study. , J Thromb Haemost 12(4), 497-504.
- 34.ZX Geng Y DI, Sun X L, Su A Y, Guo K, Li Q. (2019) Clinical Significance of Platelet Membrane Glycoprotein GPIIb/IIIa in Diagnosis and Treatment of Immune Thrombocytopenia. Zhongguo shi yan xue ye xue za zhi. 27(4), 1241-1245.
- 35.Ejaz A, Radia D. (2019) Diagnosis and management of primary immune thrombocytopenia in adults. , Br J Hosp Med 80(4), 54-57.
- 36.Matzdorff A, Wörmann B. (2018) Diagnostik und Therapie der Immunthrombozytopenie. , DMW - Dtsch Medizinische Wochenschrift 143(15), 1076-1081.
- 37.Červinek L. (2018) Diagnosis and treatment of immune thrombocytopenia. http://www.ncbi.nlm.nih.gov/pubmed/30193522. AccessedFebruary11,2020 , Vnitr Lek 64(5), 526-529.
- 38.Lucchini E, Zaja F, Bussel J. (2019) Rituximab in the treatment of immune thrombocytopenia: what is the role of this agent. in 2019? Haematologica 104(6), 1124-1135.
- 39.Cines D B, Bussel J B, Liebman H A, Luning Prak ET. (2009) The ITP syndrome: pathogenic and clinical diversity. , Blood 113(26), 6511-6521.
- 41.Wei Y, Ji X, Wang Y. (2016) High-dose dexamethasone vs prednisone for treatment of adult immune thrombocytopenia: a prospective multicenter randomized trial. , Blood 127(3), 296-302.
- 42.Guidry J A, George J N, Vesely S K, Kennison S M, Terrell D R. (2009) Corticosteroid side-effects and risk for bleeding in immune thrombocytopenic purpura: patient and hematologist perspectives. , Eur J Haematol 83(3), 175-182.
- 43.Ammann E M, Haskins C B, Fillman K M. (2016) Intravenous immune globulin and thromboembolic adverse events: A systematic review and meta-analysis of RCTs. , Am J Hematol 91(6), 594-605.
- 44.Laosombat V, Wiriyasateinkul A, Wongchanchailert M. (2000) Intravenous gamma globulin for treatment of chronic idiopathic thrombocytopenic purpura in children. http://www.ncbi.nlm.nih.gov/pubmed/10710885. AccessedFebruary13,2020 , J Med Assoc Thai 83(2), 160-168.
- 45.Lazarus A H, Crow A R. (2003) Mechanism of action of IVIG and anti-D. in ITP. Transfus Apher Sci 28(3), 249-255.
- 46.RxList.WinRho SDF ([Rh (D) Immune Globulin Intravenous (Human)] (WinRho SDF) Solution for Injection): Uses, Dosage, Side Effects, Interactions. , Warning. https://www.rxlist.com/winrho-sdf-drug.htm.Published2019. AccessedFebruary11,2020
- 47.Tarantino M D, Bussel J B, Cines D B. (2007) A closer look at intravascular hemolysis (IVH) following intravenous anti-D for immune thrombocytopenic purpura (ITP). , Blood 109(12), 5527-5527.
- 48.Cooper K L, Fitzgerald P, Dillingham K, Helme K, Akehurst R. (2012) Romiplostim and eltrombopag for immune thrombocytopenia: Methods for indirect comparison. , Int J Technol Assess Health Care 28(3), 249-258.
- 49.Chapin J, Lee C S, Zhang H, Zehnder J L, Bussel J B. (2016) Gender and duration of disease differentiate responses to rituximab–dexamethasone therapy in adults with immune thrombocytopenia. , Am J Hematol 91(9), 907-911.
- 50.Zaja F, Baccarani M, Mazza P. (2010) Dexamethasone plus rituximab yields higher sustained response rates than dexamethasone monotherapy in adults with primary immune thrombocytopenia. , Blood 115(14), 2755-2762.
- 51.Thai L-H, Mahévas M, Roudot-Thoraval F. (2016) Long-term complications of splenectomy in adult immune thrombocytopenia. , Medicine (Baltimore) 95(48), 5098-10.
- 52.Guan Y, Wang S, Xue F. (2017) Long-term results of splenectomy in adult chronic immune thrombocytopenia. , Eur J Haematol 98(3), 235-241.
- 53.KKW Wang, Charles C, Heddle N M, Arnold E, Molnar L et al. (2014) Understanding why patients with immune thrombocytopenia are deeply divided on splenectomy. , Heal Expect 17(6), 809-817.
- 54.Chaturvedi S, Arnold D M, McCrae K R. (2018) Splenectomy for immune thrombocytopenia: Down but not out.
- 55.Supe A, Parikh M, Prabhu R, Kantharia C, Farah J. (2009) Post-splenectomy response in adult patients with immune thrombocytopenic purpura. , Asian J Transfus Sci 3(1), 6.
- 56.Kojouri K, Vesely S K, Terrell D R, George J N. (2004) Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. , Blood 104(9), 2623-2634.
- 57.Thanarajasingam G, Vaidya R, Erie A, Wolanskyj A P. (2011) Accessory splenectomy for refractory immune thrombocytopenic purpura. , Am J Hematol 86(6), 520-523.
- 58.Neunert C, Terrell D R, Arnold D M. (2019) guidelines for immune thrombocytopenia. , American Society of Hematology 3(23), 3829-3866.
- 59.Rattanathammethee T, Theerajangkhaphichai W, Rattarittamrong E. (2017) The efficacy of colchicine and dapsone combination therapy in relapsed immune thrombocytopenia. Hematol Rep. 9(1), 22-27.
- 60.Kuter D J, Gernsheimer T B. (2009) Thrombopoietin and Platelet Production in Chronic Immune Thrombocytopenia. , Hematol Oncol Clin North Am 23(6), 1193-1211.
- 61.Kuter D J, Rummel M, Boccia R.Romiplostim or Standard of Care in Patients with Immune Thrombocytopenia. , N Engl J Med 363(20), 1889-1899.
- 62.Cheng G, Saleh M N, Marcher C. (2011) Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. , Lancet 377(9763), 393-402.
- 63.Al-Samkari H, Kuter D J. (2019) Optimal use of thrombopoietin receptor agonists in immune thrombocytopenia. , Ther Adv Hematol 10, 204062071984173.
- 64.Zhang L, Zhang M, Du X, Cheng Y, Cheng G. (2018) . Eltrombopag Plus Pulsed Dexamethasone As First Line Therapy for Subjects with Immune Thrombocytopenic Purpura (ITP). Blood.132(Supplement1): 733-733.
- 66.Al-Samkari H, Kuter D J. (2018) Thrombopoietin level predicts response to treatment with eltrombopag and romiplostim in immune thrombocytopenia. , Am J Hematol 93(12), 1501-1508.
- 67.Kuter D J, Meibohm A, Lopez A. (2014) TPO concentrations and response to romiplostim. , Am J Hematol 89(12), 1155-1156.
- 68.Kuter D J, Bussel J B, Lyons R M. (2008) Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. , Lancet 371(9610), 395-403.
- 69.Bussel J B, Provan D, Shamsi T. (2009) Effect of eltrombopag on platelet counts and bleeding during treatment of chronic idiopathic thrombocytopenic purpura: a randomised, double-blind, placebo-controlled trial. , Lancet 373(9664), 641-648.
- 70.Kuter D J, Bussel J B, Newland A. (2013) Long-term treatment with romiplostim in patients with chronic immune thrombocytopenia: safety and efficacy. , Br J Haematol 161(3), 411-423.
- 71.Newland A, Godeau B, Priego V. (2016) Remission and platelet responses with romiplostim in primary immune thrombocytopenia: Final results from a phase 2 study. , Br J Haematol
- 72.Steurer M, Quittet P, Papadaki H A. (2017) A large observational study of patients with primary immune thrombocytopenia receiving romiplostim in European clinical practice. , Eur J Haematol 98(2), 112-120.
- 73.DasGupta R K, Levine L, Wiczer T, Cataland S. (2019) Initial romiplostim dosing and time to platelet response in patients with treatment refractory immune thrombocytopenia. , J Oncol Pharm Pract 25(3), 567-576.
- 74.Al‐Samkari H, Kuter D J. (2018) An alternative intermittent eltrombopag dosing protocol for the treatment of chronic immune thrombocytopenia. , Br J Clin Pharmacol 84(11), 2673-2677.
- 75.Afdhal N H, Giannini E G, Tayyab G. (2012) . Eltrombopag before Procedures in Patients with Cirrhosis and Thrombocytopenia. N Engl J Med 367(8), 716-724.
- 76.Bussel J B, Kuter D J, Aledort L M. (2014) A randomized trial of avatrombopag, an investigational thrombopoietin-receptor agonist, in persistent and chronic immune thrombocytopenia. , Blood 123(25), 3887-3894.
- 77.Rastetter W, Molina A, White C A. (2004) Rituximab: Expanding Role in Therapy for Lymphomas and Autoimmune Diseases.Annu Rev Med. 55(1), 477-503.
- 79.US.National Library of Medicine. RITUXAN- rituximab injection, solution Genentech,Inc.AccessedFebruary11,2020.
- 80.Marangon M, Vianelli N, Palandri F. (2017) Rituximab in immune thrombocytopenia: gender, age, and response as predictors of long-term response. , Eur J Haematol 98(4), 371-377.
- 81.Moore D C, Gebru T, Muslimani A. (2019) Fostamatinib for the treatment of immune thrombocytopenia in adults. , Am J Heal Pharm 76(11), 789-794.
- 82.Bussel J, Arnold D M, Grossbard E. (2018) Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: Results of two phase 3, randomized, placebo-controlled trials. , Am J Hematol 93(7), 921-930.
- 83.Bussel J B, Arnold D M, Boxer M A. (2019) Long‐term fostamatinib treatment of adults with immune thrombocytopenia during the phase 3 clinical trial program. , Am J Hematol 94(5), 546-553.
- 85.Ozcelik F, Oztosun M, Arslan E. (2012) A Useful Method for the Detection of Ethylenediaminetetraacetic Acid- and Cold Agglutinin-Dependent Pseudothrombocytopenia. , Am J Med Sci 344(5), 357-362.