
Cardiorenal Signaling Pathways in Heart Failure: Good and Bad News
Abstract
This article reviews heart–kidney signaling in heart failure, contrasting adaptive and maladaptive pathways. It discusses neurohormonal activation, fibrosis, and emerging therapeutic targets.
Author Contributions
Academic Editor: Prasenjit Guchhait, Associate Professor of Biotechnology
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2016 Martin Rosas-Peralta, et al
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
During the past half-century, the advances in the prevention, diagnosis, and management of cardiovascular disease (CVD) have been nothing short of spectacular. Age-adjusted CVD related deaths have declined by about two-thirds in industrialized nations1 however, in developing countries still increasing. Nevertheless, Heart failure (HF) is a notable exception to these encouraging trends. Indeed, after normal delivery, it is the most common cause of hospitalization. Annual hospital discharges in patients with a primary diagnosis of HF have risen steadily since 1975, and now exceed 1 million discharges per year, although they may at last be leveling off or actually decreasing, in the United States. In Europe, hospitalizations for HF are clearly declining. HF is primarily a disease of the elderly that affects about 10% of men and 8% of women over the age of 60 years, and its prevalence rises with age and has risen overall. In the United States, patients with a primary diagnosis of HF now make >3 million physician visits per year. The direct and indirect costs of HF in the United States are staggering; in 2010 they were estimated to be US $39.2 billion. The estimated lifetime cost of HF per individual patient is $110,000/year (2008 US dollars), with more than three-fourths of this cost consumed by in-hospital care1
Survival after a diagnosis of HF has improved during the past 30 years; the age-adjusted death rate has declined, and the mean age at death from HF has risen. However, despite these modest improvements, the 5-year mortality is still approximately 50% worse than that of many cancers. Among Medicare patients, 30-day mortality is 10% to 12%, and the 30-day readmission rate after hospital discharge is 20% to 25%1
Cardiorenal syndrome (CRS) is the umbrella term used to describe clinical conditions in which cardiac and renal dysfunctions coexist. Much has been written on this subject, but underlying pathophysiological mechanisms continue to be unraveled and implications for management continue to be debated. Mortality is increased in patients with heart failure (HF) who have a reduced glomerular filtration rate (GFR).Patients with chronic kidney disease have an increased risk of both atherosclerotic cardiovascular disease and HF, and cardiovascular disease is responsible for up to 50 percent of deaths in patients with renal failure1, 2, 3. Acute or chronic systemic disorders can cause both cardiac and renal dysfunction. The pathophysiological pathways have common neurohormonal links such as renin-angiotensin-aldosterone system, natriuretic peptides, oxidative stress and endothelial dysfunction. This review focuses on this issue as an important and common medical situation that the physicians should always keep in mind.
Heart failure (HF) is a condition in which the heart pump is unable to provide blood supply suitable to various organs and tissues, as well as to eliminate harmful residual products. Thus HF, with its mosaic of signs and symptoms is really defined as a syndrome. In this sense, two pathognomonic symptoms are shortness of breath and fatigue, with secondary congestion, renal retention of sodium and water with elevated venous pressure favoring the transudation of intravascular fluid in the interstitial space.
Most often, older people who have HF have many risk factors that contribute to the development of this syndrome, such as hypertension, diabetes mellitus, kidney disease, obesity, COPD and sleep apnea1, 2
The severity of the clinical manifestations of HF is variable; however, the progressive condition that can have recurrent exacerbations and requires constant therapeutic interventions is defined as chronic heart failure (CHF), while a gradual or sudden quick start requiring urgent treatment is acute HF (AHF)2
We consider that a concept more contemporary and emerging for HF which goes beyond the concept of retention of sodium and water, and congestion is the real justification for this review. What is emerging is a concept of a systemic syndrome in which multiple deleterious cellular pathways are activated by mechanical and humoral mediators known and unknown.
Similarly, renal failure is clearly linked with increased adverse cardiovascular outcomes. Almost 44% of deaths in patients with end-stage renal failure (ESRF) are due to cardiovascular diseases, and a 2006 meta-analysis indicated that patients with ESRF are more likely to die from cardiovascular causes than from renal failure itself2 Death from cardiovascular causes is 10–20 times more common in patients with chronic renal failure than in matched segments of the general population. Half of patients commencing hemodialysis will suffer a myocardial infarction within the following two years, and mortality in this patient population is high. Increased myocardial mass (i.e., left ventricular hypertrophy)—which increases myocardial oxygen demand—is increased in mild-to-moderate as well as more advanced stages of renal failure.
In this sense, several cascade mechanisms result in remodeling of tissue in the heart and kidney, and probably in other systems of the body, leading inexorably to pro-fibrotic and mechanisms of apoptosis in the final phase of HF. In fact, it is in this context circulating biomarkers that can be valuable diagnostic and prognostic tools of disease as well as serve as biochemical factors of protection or deleterious enhancers of HF.
Heart failure with preserved ejection fraction (HFpEF) and with reduced ejection fraction (HFrEF)
Although each cell type and heart Chamber can potentially be involved in the beginning of the HF, there is often to top left ventricular (LV) dysfunction linked to a lifting of the pressures of filling of the LV3, 5, 6 (Diastolic Disfunction).Figure 1.
Thus, a bad secondary adaptation to the loss of the muscle due to myocardial infarction, the reduction of an increase in afterload or contractility due to idiopathic cardiomyopathy as result of hypertension they generate at the time a low heart beat volume and increased tension of the LV wall with performance altered preload.
Figure 1.Deflection curve of volume/pressure ratio in heart failure by diastolic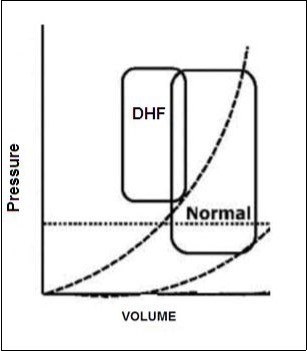
In the last decade it has been described that when the LV ejection fraction (EF) is reduced (≤40%), is a condition called "systolic dysfunction", what defines the IC as "IC with reduced ejection fraction" (HFrEF). When LVEF is ≥50%, but have concomitant impaired relaxation of the left ventricle, a condition called "diastolic dysfunction", along with the presence of pathognomonic signs and symptoms of IC, is classified as "IC with preserved ejection fraction" (HFpEF).
Individuals with heart failure and a faith between 40% and 50% are considered as part of an intermediate group (gray area), situation which we have also known as HF with systolic dysfunction in transit3. In addition, LV diastolic dysfunction may also be present in the own HFrEF4Interestingly, HFrEF and HFpEF may be differentiated according to the phenotype of the patient. HFpEF subjects, compared with HFrEF, are often older women, with a higher body mass index, increased prevalence of diabetes, atrial fibrillation and a history of high blood pressure5. The estimated prevalence of HFpEF among subjects with IC is approximately 50%6, 7. So is today already recognized as pandemic, by its close relationship with factors of cardiovascular risk such as hypertension, diabetes, Dyslipidemia, obesity, COPD and sleep apnea.
The mortality rate may be higher in patients with HFrEF, however, the high prevalence of HFpEF leads the largest absolute number of deaths, then still numerically greater epidemiological impact HFpEF as cause of death6. Patients with HFpEF die mainly due to cardiovascular causes; nevertheless, they also have a higher incidence of non-cardiovascular mortality compared to HFrEF. The above probably related to the associated comorbidities5, 8. The structural characteristics of the HFrEF and HFpEF are markedly different. In HFrEF, the LV is dilated with hypertrophic walls9. Histologically, fibrosis is present, cardiac myocytes are elongated and they have a smaller diameter than in HFpEF and also its inner myofibrillar density is reduced. In addition, the myocytes are less rigid in comparison with HFpEF.
HFpEF, LV cavity typically has a normal volume and the walls are hypertrophic. See Figure 2. Histological examination shows the deposition of collagen and cardiomyocytes larger, stiffer than in HFrEF4. Despite the different cardiac structure and function, HFrEF and HFpEF hemodynamic patterns share similarities as well as differences. The clinical symptoms, renal dysfunction, neurohormonal activation, response to exercise and clinical findings may overlap10 However, increased ventricular and vascular stiffness can play a greater role than a real volume overload, in HFpEF compared with HFrEF. Thus, these two forms of IC are two well differentiated entities, with different pathophysiology and therapeutic approaches accordingly.
Figure 2.Model of concentric left ventricular hypertrophy, common in HFpEF.
Comorbidities HFpEF-associated lead to endothelial dysfunction, a condition characterized by the alteration of the "Endothelial Activation" and endothelium-dependent vasodilation, referring to a state where the endothelium loses its physiological properties and moves to a state pro-inflammatory, pro-coagulator and vasoconstrictor.
It is possible that HFpEF can evolve to HFrEF and, therefore, both conditions can be considered "the ends of a single disease"11 The treatment and prognosis of cardiovascular disease, has significantly improved in recent decades. However, the increase in mortality and hospitalization rates remain elevated in patients with HF (5). American guidelines for the treatment of the HF3 are more clear and simple to HFrEF; However, missing consensus HFpEF management8.
More recently, the PARAMOUNT Study12 was a phase II clinical trial that tested the effectiveness of a novel compound created by the combination of an ARA2, Valsartan, and an inhibitor of Neprilisina (Sacubitril, AHU377). Neprilisina (metal-neutral endopeptidase24, 11 is one of the most important enzymes responsible for the degradation of natriuretic peptides (PNs) among others. The name of the first drug in its class (Angiotensin Receptor Neprilysin inhibitor, RNAI) is LCZ696. The strategy behind this complex molecule is based on the effect of two different, both pathways important in the pathogenesis of HFrEF and HFpEF: Renin-angiotensin-aldosterone system-(RAAS) and the Neprilisina system8.
This new drug is added to the basal therapy in n = 301 patients HFpEF compared with subjects with initial treatment only valsartan. The results of this study showed a greater reduction in the levels of NT-proBNP in the group treated with LCZ696 compared with controls; however, this difference was not present after 36 weeks of observation. Moreover, this reduction in the levels of NT-proBNP appears to translate into better clinical results. LCZ696 has recently been added to standard therapy in chronic HFrEF patients symptomatic: PARADIGM-IC 13, 14. This study was arrested so early (March 2014 instead of October 2014) due to the effect on mortality in subjects that LCZ696 compared with subjects in standard therapy with an ACE Inhibitor (Enalapril). LCZ696 compared with enalapril also reduced the risk of hospitalization for IC and significantly improved the symptoms of HF.
These impressive results have been obtained using a drug that targets at the same time for RAAS and the NP system supporting a favorable effect and increase of the combination of the two molecules. LCZ696 may change the therapeutic strategy and survival to long-term HFrEF patients 15; however, topics selected for this landmark study is that they had an LVEF ≤35% and had to tolerate a dose of 10 mg twice a day of enalapril before being considered to take LCZ696. The translation into clinical practice may require careful considerations.
Another essential essay is TOPCAT that tested the efficacy of spironolactone in patients with HFpEF16. In this case the researchers selected patients with an EF ≥45%, in the Americas, as well as Russia and Georgia, and reported that spironolactone treatment did not reduce the primary composite outcome death from cardiovascular causes, aborted cardiac arrest or hospitalization for treatment of heart failure. It is important because of RAAS blockade is crucial for survival outcome in HFrEF but not in HFpEF17.
In conclusion, the treatment of the HF and, particularly, HFpEF remains a challenge that certainly warrants new therapeutic approaches to alternative and innovative, in the acute setting, as well as in CHF.
Key element in the pathogenesis of HFpEF: Endothelial dysfunction
The cardiac endothelium comprises the endothelial cells of the coronary microvasculature, endocardium, and the capillary intramyocardial. Cardiomyocytes are within the coronary microvascular network to maximum 3 microns in endothelial cells. This specific anatomical layout allows not only the adequate blood supplies, but also facilitates two-way communication between cells. The relevance of the cardiac endothelium in cardiac function follows from the work pioneers who demonstrated in vitro that the cardiac endocardial surface modulates cardiac muscle performance.
By coronary infusion of the substance P which leads to acute modulation of left ventricular function (LV), provided evidence later that the cardiac endothelium is important for cardiac contractile function in human beings potentially through the release of paracrine factors including ON, endothelin-1, natriuretic peptides, cytokines, and others.
Thus, emphasizes the importance of the cardiac endothelium function of cardiomyocytes, i.e. the cardiac endothelium and consequently Endothelial dysfunction or damage affects cardiomyocytes as well as cardiac fibroblasts and the intracardiac neurohormonal activation. The impact of the cardiac endothelium at these different heart cells along with complex interactions (bi-directional) emphasizes the relevance of the Endothelial dysfunction in the pathogenesis of HFPEF.
Endothelial-to-Mesenchymal Transition (EndMT)
A very relevant aspect is remember deprivation nitric oxide helps transition endothelial-mesenchymal, a process whereby endothelial cells become a type of cell mesenchymal, which can give rise to fibroblasts. EndMT is induced by inflammatory factors (TGF-ss and TNF-α), oxidized LDL and by age. Also the EndMT contributes to cardiac fibrosis during chronic pressure overload.
Recently, specific endothelial expression of endothelin-1 and NOX2 (NADPH oxidase-2) have shown that they induce EndMT in experimental models of diabetes mellitus and cardiac hypertrophy and fibrosis induced by Ang II, respectively. Interestingly, the activation Ang II-induced endothelial NOX2 is associated with isolated in the absence of systolic dysfunction, diastolic dysfunction.
In summary, these findings suggest that activation by endothelial dysfunction and inflammation not only, also induce the much less appreciated EndMT, as important mechanisms that contribute to cardiac fibrosis and the development of HFPEF. Cardiac fibroblasts in turn may trigger more cardiac inflammation in a multimodal way. This complex interaction between them cells endothelial, cells inflammatory and fibroblasts heart could explain why those strategies unidirectional of counter inflammation or fibrosis have failed until now to block the process fibrotic e indicates the need of new strategies with effects immunomodulatory more spacious. In addition, emphasis is on the need to diagnose and treat HFPEF in an early stage of its pathogenesis, where the vicious cycle of inflammation and fibrosis, which leads to chronic inflammation, might even be repealed.
Neurohormonal Activation in HF.
Despite different pathophysiology, HFrEF and HFpEF share the activation of three main systems neurohormonales: NP system, the sympathetic nervous system (SNS) and RAAS. Neurohormonal activation has laid the foundations in the field of biomarkers of IC. The initial phase of HF is usually asymptomatic. Cardiomyocytes that distended in the heart failure secrete natriuretic peptides mainly from the Atria18 to reduce the hemodynamic impairment secondary to vasoconstriction and retention of sodium by the SNS and RAAS1.
More specifically, the SNS increases inotropic function and peripheral vasoconstriction19, while RAAS maintains and expands intravascular volume and renal perfusion by vasoconstriction in the kidney and tubular sodium reabsorption. Natriuretic peptides of human system consists of three structurally similar but genetically different hormones: natriuretic peptide, atrial natriuretic peptide (ANP) type B (BNP) and natriuretic peptide type C (NPC). ANP and BNP are mainly synthesized in the heart while the NCP is mainly produced by the endothelium and the kidney. ANP and BNP acting through a receiver of membrane associated guanylate Cyclase (GC-A). The NCP binds preferentially to the receiver of particulate guanylate Cyclase (GC-B) B.
There is a third receiver for the natriopeptides receptor C (NPR-C), which can also have proliferative action on cardiomyocytes and anti-fibrotic actions in cardiac fibroblasts20,21 properties. Only GC-A & GC-B, after linking its specific peptides, produced the second Messenger Guanosine cyclic 3′, 5′-monophosphate (cGMP). CGMP, importantly, that it is involved in several cardiovascular actions which imply the Suppression of cell proliferation, inhibition of Platelet Activation and reduce inflammation8, 22 as well as the preservation of myocardial function and its structure23. Plasma natriuretic peptides (PNs) elevation and, subsequently, cGMP levels can be seen as a compensatory response to the initial cardiovascular adaptation in the IC. The PNs have numerous and notable actions including natriuresis24, inhibition of the synthesis of aldosterone25 and increase vasodilation26.
The PNs are not the only contributors to the increased levels of cGMP. The nitric oxide (no) to act through guanylate Cyclase soluble, another receptor guanylate Cyclase, through the production of cGMP can modulate inflammation27, 28, 29) Endothelial dysfunction and myocardial contractility. However, ON bioavailability may be reduced in IC30, contributing to a State of relative deficiency of cGMP. The PNs, along with ON, try to compensate for hemodynamic dysfunction and characterized the initial stage of IC, through the activation of cGMP. However, the NHS, that releases catecholamines, induced opposite effects, and can also turn directly to RAAS31. Peripheral vasoconstriction, including vasoconstriction of the renal arteries, induced by the SNS can also lead to hypoperfusion followed by release of Renin from the glomerular kidney and consequently increases the activation of the RAAS.
Directly or indirectly, there are greater renal involvement, with an increase of angiotensin II (Ang II) and finally the levels of aldosterone. It is important that the kidney plays a crucial role during the IC, ultimately leading to the retention of sodium and water through the activation of the Mineralocorticoid receptors in the distal nephron32. In addition, Ang II may stimulate the release of norepinephrine, keeping a vicious circle between the SNS and RAAS33. During the progression of the disease, systemic vasoconstriction and increased intravascular volume secondary to renal retention of salt and water are harmful not only for the function cardiorenal but also changes in its structure. RAAS and SNS actions overwhelm the beneficial effects of PNs-ON/cGMP leading to symptomatic HF, increased re-hospitalization and increased risk of death.
It is important to underline that these complex interactions are present in both heart failure, acute and chronic, although with different levels of severity. On the other hand, after a myocardial infarction, aldosterone is also in its maximum plasma concentration34. In fact, EPHESUS is an essay clinical pivot in which them researchers tested the efficacy of a blocker of the receiver Mineralocorticoid, eplerenone, given after infarction acute of myocardial complicated by the dysfunction ventricular left and IC. This trial showed that the addition of this drug to the therapy medical optimal reduces the morbidity and mortality35,46. In addition, this medication has shown a benefit of survival early. Significant reduction of the mortality was fined 30 days after the randomization in patients with HFrEF36).
Therefore, the role of aldosterone in the pathogenesis of cardiovascular diseases clearly goes beyond heart failure and hypertension.
Cardiorenal Syndrome.
Originally specified by Dr. Eugene Braunwald, the key symptoms of CHF (dyspnea and fatigue) are consequence of the retention of salt and water 5 by the kidney. Therefore, the interaction between the heart and the kidney is being a component crucial of the CHF. Finally, HF becomes both in acute conditions or chronic state in a real cardiorenal syndrome where the glomerular filtration rate is inadequate and retention of sodium and water prevail with refractoriness to the endogenous natriuretic peptides, and diuretics. When the heart begins to fail, the yuxtaglomerular cells secrete Renin.
Renin cleaves to angiotensinogen,Figure 3 produced and released into the circulation by the liver into angiotensin I. ACE, widely present along the vasculature and into the renal tubules, cleaves angiotensin I giving Ang II. Ang II, is also a potent vasoconstrictor, activates the synthesis of aldosterone and its release of the zona glomerulosa of the adrenal cortex, with secondary increase in circulation of the same32. Aldosterone binds to the receptor of mineralocorticoids in the epithelial cells of the duct collector of the nephron, inducing the excretion of potassium and sodium retention (RM).
Figure 3.Classical and non-classical enzymatic production of angiotensin pathways
This action is counter-regulated by the natriuretic effect of cardiac ANP and BNP that is at least at the beginning, effective. However, with the evolution of the HF, the actions of protection of PNs/cGMP are lost, leading to the progression of the disease. The volume overload induces more loss of cardiomyocytes by stress and cell apoptosis. Also, start with the accumulation of extracellular matrix pro-fibrotic and inflammatory processes. The remodeling, hypertrophy, fibrosis and death with regeneration reduced myocardial37 eventually irreversible heart damage and end-stage of HF. In parallel, the kidney undergoes adaptation and remodeling initially compensatory and finally deleterious with loss of renal function.
Renal blood flow is reduced, and/or the reduced venous return can result in renal congestion38, 39, 40) with secondary increase in renal interstitial pressure41. HFpEF and HFrEF are strongly associated with renal dysfunction39, 42). This finding endorses the definition of Cardiorenal syndrome. Figure 4.
As in the heart, renal perfusion impaired long-term induces local routes in fibrosis and consequent reduction in the glomerular function and tubular with eventual damage to the parenchyma (chronic kidney disease, CKD).
Figure 4.Types of Cardiorenal syndrome. Note that this classification stages depend on if it is acute failure or chronic heart and if the insult is initially cardiac or renal.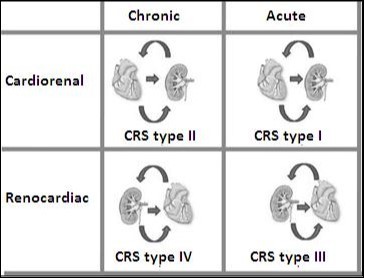
This is the picture of the cardiorenal syndrome with high risk of death and rehospitalization. This syndrome has been classified by the Working Group of the initiative for the quality of dialysis, which proposes a 5 subtypes classification scheme academic way. This classification of 5 items is based on: a) if the principal organ of the dysfunction is the heart, kidney, or a third independent process that affects both the bodies and b) the acute or chronic nature of the disease. Table 1 summarizes the current 5 types of CRS.
This strong and complex intercommunication between the heart and the kidney is bidirectional. Therefore, the kidney may even be the first body to start the IC. This concept is supported by earlier clinical studies that have shown even mild renal impairment as a factor that contributes to an increased cardiovascular risk43, 44). In a recent article45) showed that slight ERC, produced by uninephrectomy (UNX), was in early cardiac fibrosis with mild impairment diastolic and systolic function preserved in rats.
Table 1. Classification of Cardiorenal Syndrome.| CRS types | Hallmarks | Clinical sceneries |
| CRS type 1(acute CRS) | The rapid deterioration of heart function that leads to AKI | Acute Decompensated HF, AMI with cardiogenic shock, acute valvular insufficiency |
| CRS type 2 (chronic CRS) | Chronic alterations in cardiac function leading to chronic progressive renal disease | Chronic HF (activation of the RAAS and SNS long-term, chronic hypoperfusion) |
| CRS type 3 (acute renocardiac syndrome) | Primary worsening of renal function leading to acute cardiac dysfunction. Overload of fluids, electrolyte disturbances, the accumulation of myocardial depressants factors, neurohormonal activation, and systemic inflammation have been postulated to lead to cardiac dysfunction | Acute kidney injury (uremia causing deterioration of contractility, hyperkalaemia cause arrhythmias, the volume overload causing pulmonary edema)) |
| CRS type 4 (chronic renocardiac syndrome) | Chronic kidney disease primary contributing to decreased cardiac function and an increased risk of cardiovascular events | The chronic kidney disease that leads to the HVI, coronary artery disease, and diastolic dysfunction |
| CRS type 5 (secondary CRS) | Whether the presence of concomitant cardiac and renal dysfunction due to acute or chronic systemic disorders | Diabetes mellitus, vasculitis, amyloidosis, sepsis, cardiogenic shock |
The consensus report: Acute Dialysis Quality Initiative. European Heart Journal (2010) 31, 703-711. CRS, Cardiorenal syndrome; AKI, acute Kidney injury; AMI, acute myocardial infarction; RAAS, Renin-angiotensin-aldosterone system; LVH, Left ventricular hypertrophy
These results were independent of blood pressure, sodium, water retention or the activity of aldosterone. In addition, this connection of the kidney - heart in early CKD could involve at least two pathways of genes in the heart: pathways of TGF-β and apoptosis. These important findings support the hypothesis that impaired renal function is associated with release of renal cellular or humoral factors that contribute to changes in the structure and function of the myocardium.
Arginine vasopressin (AVP), also known as antidiuretic hormone (ADH), is synthesized by the hypothalamus and stored in the neurohypophysis. This hormone induces the reabsorption of water by the nephron, ultimately increases the intravascular volume48. AVP is also part of the neurohormonal activation during IC.
Adrenomedullin (ADM) is a peptide vasodilator first found in cells of pheochromocytoma, but has subsequently been found in various organs such as the heart, kidney, lung, smooth muscle cells, and endothelium5, 49, 50. LOA ADM levels are elevated during IC to reduce preload and afterload.
Cortisol is produced in the zona fasciculata of the adrenal cortex. This hormone glucocorticoid, locally not completely inactivated the enzyme 11β-hydroxysteroid dehydrogenase type 2 (11 βHSD2), can induce sodium and water retention during IC, using the link to the RM in the kidney51, 52. In IC, cortisol levels are higher than in subjects without IC53.
The activation of the endothelium, the extracellular matrix, cells of the immune system and the reaction of oxidative stress also contribute to the deadly progression of a phase offset to a lopsided and symptomatic stage IC. Specifically, Endothelins, released by endothelial cells, induces vasoconstriction and release of aldosterone54, 55. Cytokines such as TNF-α, IL-1, IL-6, inflammatory proteins and other molecules are produced by lymphocytes, macrophages, endothelial cells, and many other tissues under conditions of stress. The injured myocardium can also secrete Pro-Inflammatory Cytokines. All of these factors culminate in progression to ICC56.
More recently, in the systemic circulation of the IC holder, it is also possible to detect matrix proteinases, the enzymes involved in the metabolism of collagen. Its presence indicates extracellular matrix remodeling and active processes fibrotic5. Indeed, it has shown changes in biomarkers of synthesis and degradation of collagen in patients with congestive heart failure and left ventricular systolic dysfunction after acute myocardial infarction. This remodeling of extracellular matrix was attenuated by the antagonist of aldosterone57. In addition, the ST2 soluble receptor of interleukin released in response to the tension of myocardial58, 59, 60 and galectin-3, inducing mediator of macrophages derived from the synthesis of collagen 61, are also measured to evaluate the severity/prognosis of IC. Soluble ST2 plays a role in the cardio-protective stress response, while galectin-3 is involved in the fibrotic heart process.
Therefore, both ST2 soluble and galectin-3 together with the peptidases of collagen, are currently widely used, demarcating the important concept of the volume control in interaction with extracellular collagen during IC.
The ischemic injury could characterize the progression of IC, causing release of peptides specific cardiac, such as troponins in the bloodstream47.
Inflammation and Endothelial dysfunction, participate directly in the activation of fibroblasts responsible for excess remodeling and fibrosis of the extracellular matrix. figure 5
Figure 5.Direct and indirect way of activation of fibroblasts by monocyte-macrophages.
In the IC has been also reported that there may be a secondary hyperparathyroidism. Specifically, parathyroid hormone (PTH) is released from the parathyroid glands when there is hypocalcemia. Therefore, calcium homeostasis may be impaired in IC, as well as ERC. In addition, PTH has shown direct pleiotropic effects in cardiomyocytes62. Another connection between IC and altered bone metabolism is the version of growth factor fibroblastic 23 (FGF-23) from the Osteocyte63.
The growth of fibroblasts, FGF 23, factor has direct actions on myocardial hypertrophy. In short, all of these factors are considered valid biomarkers of IC. However, to date, BNP and N-terminal of the Pro-hormone (NT-proBNP) are the only ones recommended by the American Heart Association for the diagnosis of IC. Besides BNP, biomarkers of damage (troponin) myocardial and cardiac fibrosis (ST2 soluble and galectin-3) may be considered for the risk stratification of additive in ICA3.
An ideal biomarker should then allow the diagnosis, to help in the monitoring of the treatment and prognosis of a specific disease, in addition to being highly sensitive, specific, reliable and standardized, regardless of age, sex, and anthropometric parameters of the subject64 IC, despite continuous research and identification of new biomarkers, questions and challenges remain. Biomarkers are part of the major pathways of the disease and provide a powerful rational for us to monitor specific pathological conditions, such as IC.
Then one could speculate that the use of biomarkers to track the disease might not totally be an optimal strategy65. in the IC, the use of ARA2, and, recently, the new class of drugs ARNIs are examples of therapeutic strategies for biomarkers. The effectiveness of these drugs, especially in HFrEF is unquestionable.
Nevertheless endothelial dysfunction and increased oxidative stress seem to be important pathophysiologic processes associated with the pro-inflammatory state of HFpEF and HFrEF66. Increase of oxidative stress and endothelial dysfunction has been described in both HFpEF and HFrEF, but is also implicated in other diseases, including diabetes, hypertension, both highly prevalent in HFpEF67. However, HFpEF patients demonstrated a higher prevalence of endothelial dysfunction even after matching for age, gender, diabetes and hypertension68. This suggests that increased oxidative stress and endothelial dysfunction are not merely a result of a myriad of comorbidities, but linked specifically to the pathophysiology of HF.
In a study comparing HFpEF patients with age- and sex-matched hypertensives without symptoms as well as age- and sex-matched healthy controls, global vascular function (Ea and SVRI) was not significantly different between groups, but endothelial function was impaired in both HFpEF and hypertensive subjects compared to the controls69. Interestingly, HFpEF patients had an impaired endothelial function accompanied by a reduction in exercise capacity, but hypertensive controls displayed only an impaired endothelial function, suggesting that endothelial dysfunction does not directly lead to reduced exercise capacity. However, endothelial dysfunction did predict cardiovascular events, independently of age, diabetes, hospitalization, NYHA class, E/e′, ejection fraction and BNP68 Since endothelial dysfunction is associated with prognosis in HFpEF, this could be an important pathophysiological mechanism and the pathways involved could be potential therapeutic targets.
Nitric oxide (NO) is considered a key player in endothelial dysfunction. NO promotes LV relaxation through cyclic guanosine monophosphate (cGMP)–protein kinase G (PKG) dependent and independent mechanisms. Myocardial samples from HFpEF patients displayed reduced levels of PKG activity and lower cGMP concentration, which was related to increased cardiomyocyte stiffness, demonstrated by measurement of passive tension 66. The downregulation of cGMP–PKG was likely related to low myocardial NO bioavailability, demonstrated by high nitrosative/oxidative stress assessed by immunohistochemical determination of nitrotyrosine. PKG dependent phosphorylation of the sarcomeric protein titin seems to play an important role in this process66.
High diastolic stiffness was correlated to relative hypophosphorylation of the stiff N2B titin isoform, and in HFpEF the N2BA:N2B expression ratio was decreased compared to HFrEF patients66 and69, 70). The lack of PKG in the cardiomyocyte enhances hypertrophy, as seen in experimental and clinical studies71. In patients with diabetic cardiomyopathy and concentric LV remodeling, sildenafil treatment increased myocardial PKG activity, reduced hypertrophy, and improved cardiac kinetics.
Endothelial dysfunction is not just confined to the systemic or the coronary arteries, but also includes the pulmonary arteries. Pulmonary hypertension (PH) is a common feature in HFpEF. In a community-based study, PH was present in 83% of HFpEF patients66. This population demonstrated an increased pulmonary capillary wedge pressure, however the severity of PH suggests an additional pre-capillary component contributes as well. It is possible that endothelial dysfunction plays a role in that aspect. In HFpEF patients with pulmonary hypertension, sildenafil treatment resulted in improvement of diastolic stiffness and reduction of pulmonary pressure72.
The above data suggest an important role for inflammation and inflammation-induced endothelial dysfunction in the pathophysiology of HFpEF. Targeting these processes and the pathways involved, such as cGMP–PKG, could be potentially beneficial to HFpEF patients.
Perspectives
Despite advances in science and medicine of the cardiovascular diseases, IC is still a huge burden as a public health problem. The need for new effective drugs should involve returning to the study of the pathophysiology of HFrEF and HFpEF and redefine new and different paths and biomarkers to aim with new therapies. Recognizing the activation of several hormonal systems in IC and the imbalance between PNs and RAAS, the use of natriuretic peptide receptor agonists, could be justified to increase the beneficial effects of PNs/cGMP and counteract the negative actions of Ang II and the activation of the Mineralocorticoid receptor.
However, any disease in which the majority of risk factors are modifiable, HF prevention is the first step. In addition some reports emphasize the importance of the monitoring of blood pressure, central obesity and peptide N-terminal procollagen-III to identify the first structural and functional changes in the heart, before HF stage B-C or D 73. The same group, published a proportion reduced between type III procollagen N-terminal Pro-peptide and collagen type I telopeptide as a predictive of ventricular remodeling as well as cardiovascular deaths and hospitalizations for HF, as well as BNP and cardiac LVEF74. These studies underscore the importance of biomarkers in the remodeled adverse cardiac prevention and progression of IC. In conclusion, strict monitoring of high-risk patients identified, with the use of biomarkers and modulation of their objectives, could help in the early diagnosis and therefore in a more personalized strategy to reduce the burden of this syndrome.
References
- 2.Gheorghiade M, Zannad F, Sopko G, Klein L, Pina I L et al. (2005) Acute heart failure syndromes: current state and framework for future research. , Circulation 112, 3958-68.
- 3.Yancy C W, Jessup M, Bozkurt B, Butler J, Casey Jr DE et al. (2013) ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013;128:e240–327.
- 4.L van Heerebeek, Borbely A, Niessen H W, Bronzwaer J G, Velden J van der et al. (2006) Myocardial structure and function differ in systolic and diastolic heart failure. , Circulation; 113, 1966-73.
- 6.Lam C S, Donal E, Kraigher-Krainer E, Vasan R S. (2011) Epidemiology and clinical course of heart failure with preserved ejection fraction. , Eur J Heart Fail; 13, 18-28.
- 7.Owan T E, Hodge D O, Herges R M, Jacobsen S J, Roger V L et al. (2006) Trends in prevalence and outcome of heart failure with preserved ejection fraction. , N Engl J Med; 355, 251-9.
- 8.Komajda M, Lam C S. (2014) Heart failure with preserved ejection fraction: a clinical dilemma. , Eur Heart J; 35, 1022-32.
- 10.Borlaug B A, Redfield M M.Diastolic and systolic heart failure are distinct phenotypes within the heart failure spectrum. Circulation 2011;123: 2006–13 (discussion 14).
- 11.Little W C, Zile M R. (2012) ICFEp: cardiovascular abnormalities not just comorbidities. Circ Heart Fail;. 5, 669-71.
- 12.Solomon S D, Zile M, Pieske B, Voors A, Shah A et al. (2012) The angiotensin receptor neprilysin inhibitor LCZ696 in heart failure with preserved ejection fraction: a phase 2 double-blind randomised controlled trial. , Lancet; 380, 1387-95.
- 13.McMurray J J, Packer M, Desai A S, Gong J, Lefkowitz M P et al. (2013) Dual angiotensin receptor and neprilysin inhibition as an alternative to angiotensin-converting enzyme inhibition in patients with chronic systolic heart failure: rationale for and design of the Prospective comparison of ARNI with ACEI to Determine Impact on Global Mortality and morbidity in Heart Failure trial (PARADIGM-IC). , Eur J Heart Fail; 15, 1062-73.
- 14.McMurray J J, Packer M, Desai A S, Gong J, Lefkowitz M P et al. (2014) Angiotensin–neprilysin inhibition versus enalapril in heart failure. , N Engl J Med; 371, 993-1004.
- 15.Jessup M. (2014) Neprilysin inhibition—a novel therapy for heart failure. , N Engl J Med; 371, 1062-4.
- 16.Pitt B, Pfeffer M A, Assmann S F, Boineau R, Anand I S et al. (2014) Spironolactone for heart failure with preserved ejection fraction. , N Engl J Med; 370, 1383-92.
- 17.McMurray J J, O'Connor C. (2014) Lessons from the TOPCAT trial. , N Engl J Med; 370, 1453-4.
- 18.Burnett Jr JC, Kao P C, Hu D C, Heser D W, Heublein D et al. (1986) Atrial natriuretic peptide elevation in congestive heart failure in the human. , Science; 231, 1145-7.
- 19.Triposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G et al. (2009) The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. , J Am Coll Cardiol; 54, 1747-62.
- 20.Becker J R, Chatterjee S, Robinson T Y, Bennett J S, Panakova D et al. (2014) Differential activation of natriuretic peptide receptors modulates cardiomyocyte proliferation during development. , Development; 141, 335-45.
- 21.Sangaralingham S J, Huntley B K, Martin F L, McKie P M, Bellavia D et al. (2011) The aging heart, myocardial fibrosis, and its relationship to circulating C-type natriuretic peptide. , Hypertension; 57, 201-7.
- 22.Moro M A, Russel R J, Cellek S, Lizasoain I, Su Y et al. (1996) cGMP mediates the vascular and platelet actions of nitric oxide: confirmation using an inhibitor of the soluble guanylyl cyclase. , Proc Natl Acad Sci U S A ; 93, 1480-5.
- 23.Greene S J, Gheorghiade M, Borlaug B A, Pieske B, Vaduganathan M et al. (2013) The cGMP signaling pathway as a therapeutic target in heart failure with preserved ejection fraction. , J Am Heart Assoc;2:e000536
- 24.Burnett Jr JC, Opgenorth T J, Granger J P. (1986) The renal action of atrial natriuretic peptide during control of glomerular filtration. , Kidney Int; 30, 16-9.
- 25.Richards A M. (1996) The renin–angiotensin–aldosterone system and the cardiac natriuretic peptides. , Heart; 76, 36-44.
- 26.Melo L G, Veress A T, Ackermann U, Sonnenberg H. (1998) Chronic regulation of arterial blood pressure by ANP: role of endogenous vasoactive endothelial factors. , Am J Physiol; 275, 1826-33.
- 27.Eiserich J P, Hristova M, Cross C E, Jones A D, Freeman B A et al. (1998) Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. , Nature; 391, 393-7.
- 28.Liu V W, Huang P L. (2008) Cardiovascular roles of nitric oxide: a review of insights from nitric oxide synthase gene disrupted mice.Cardiovasc Res;. 77, 19-29.
- 29.Wilkinson I B, Franklin S S, Cockcroft J R. (2004) Nitric oxide and the regulation of large artery stiffness: from physiology to pharmacology. , Hypertension; 44, 112-6.
- 30.Bhushan S, Kondo K, Polhemus D J, Otsuka H, Nicholson C K et al. (2014) Nitrite therapy improves left ventricular function during heart failure via restoration of nitric oxide-mediated cytoprotective signaling. Circ Res;. 114, 1281-91.
- 31.Jackson G, Gibbs C R, Davies M K, Lip G Y. (2000) . , ABC of heart failure. Pathophysiology. BMJ; 320, 167-70.
- 33.von Lueder TG, Sangaralingham S J, Wang B H, Kompa A R, Atar D et al.et al.Renin–angiotensin blockade combined with natriuretic peptide system augmentation:novel therapeutic concepts to combat heart failure. Circ Heart Fail 2013;6:. 594-605.
- 34.Beygui F, Vicaut E, Ecollan P, Machecourt J, E Van Belle et al. (2010) Rationale foran early aldosterone blockade in acute myocardial infarction and design of theALBATROSS trial. , Am Heart J; 160, 642-8.
- 35.Pitt B, Remme W, Zannad F, Neaton J, Martinez F et al. (2003) Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. , N Engl J Med; 348, 1309-21.
- 36.Pitt B, White H, Nicolau J, Martinez F, Gheorghiade M et al. (2005) Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. , J Am Coll Cardiol; 46, 425-31.
- 37.Vigliano C A, Cabeza Meckert PM, Diez M, Favaloro L E, Cortes C et al. (2011) Cardiomyocyte hypertrophy, oncosis, and autophagic vacuolization predict mortality in idiopathic dilated cardiomyopathy with advanced heart failure. , J Am Coll Cardiol; 57, 1523-31.
- 38.Ronco C, Haapio M, House A A, Anavekar N, Bellomo R. (2008) Cardiorenal syndrome. , J Am Coll Cardiol; 52, 1527-39.
- 39.Metra M, Cotter G, Gheorghiade M, Dei Cas L, Voors A A. (2012) The role of the kidney in heart failure. , Eur Heart J; 33, 2135-42.
- 40.Bock J S, Gottlieb S S. (2010) Cardiorenal syndrome: new perspectives. , Circulation; 121, 2592-600.
- 41.Burnett Jr JC, Knox F G. (1980) Renal interstitial pressure and sodium excretion during renal vein constriction. , Am J Physiol; 238, 279-82.
- 42.Gori M, Senni M, Gupta D K, Charytan D M, Kraigher-Krainer E et al. (2014) Association between renal function and cardiovascular structure and function in heart failure with preserved ejection fraction. Eur Heart J.
- 43.Dries D L, Exner D V, Domanski M J, Greenberg B, Stevenson L W. (2000) The prognostic implications of renal insufficiency in asymptomatic and symptomatic patients with left ventricular systolic dysfunction. , J Am Coll Cardiol; 35, 681-9.
- 44.Fried L F, Shlipak M G, Crump C, Kronmal R A, Bleyer A J et al. (2003) Renal insufficiency as a predictor of cardiovascular outcomes and mortality in elderly individuals. , J Am Coll Cardiol; 41, 1364-72.
- 45.Martin F L, McKie P M, Cataliotti A, Sangaralingham S J, Korinek J et al. (2012) Experimental mild renal insufficiency mediates early cardiac apoptosis, fibrosis, and diastolic dysfunction: a kidney–heart connection. , Am J Physiol Regul Integr Comp Physiol;302:R292 – 9.
- 46.Anand I S. (2003) Changes in brain natriuretic peptide and norepinephrine over time and mortality and morbidity in the Valsartan Heart Failure Trial (Val-HeFT). , Circulation; 107, 1278-83.
- 48.JJt Finley, Konstam M A, Udelson J E. (2008) Arginine vasopressin antagonists for the treatment of heart failure and hyponatremia. , Circulation; 118, 410-21.
- 49.Kitamura K, Kangawa K, Kawamoto M, Ichiki Y, Nakamura S et al. (1993) Adrenomedullin: a novel hypotensive peptide isolated from human pheochromocytoma. , Biochem Biophys Res Commun; 192, 553-60.
- 50.Yu C M, Cheung B M, Leung R, Wang Q, Lai W H et al. (2001) Increase in plasma adrenomedullin in patients with heart failure characterised by diastolic dysfunction. , Heart; 86, 155-60.
- 51.Whitworth J A, Mangos G J, Kelly J J. (2000) Cushing, cortisol, and cardiovascular disease. , Hypertension; 36, 912-6.
- 52.Funder J W. (2010) Minireview: aldosterone and mineralocorticoid receptors: past, present, and future. , Endocrinology; 151, 5098-102.
- 53.Anand I S, Ferrari R, Kalra G S, Wahi P L, Poole-Wilson P A et al. (1989) Edema of cardiac origin. Studies of body water and sodium, renal function, hemodynamic indexes, and plasma hormones in untreated congestive cardiac failure. , Circulation; 80, 299-305.
- 54.Lerman A, Sandok E K, Hildebrand Jr FL, Burnett Jr JC. (1992) Inhibition of endotheliumderived relaxing factor enhances endothelin-mediated vasoconstriction. , Circulation; 85, 1894-8.
- 55.Kiowski W, Sϋtsch G, Hunziker P, Mϋller P, Jonghun K et al. (1995) Evidence for endothelin-1-mediated vasoconstriction in severe chronic heart failure. , Lancet; 346, 732-6.
- 56.Anker S D. (2004) Inflammatory mediators in chronic heart failure: an overview. , Heart; 90, 464-70.
- 57.Iraqi W, Rossignol P, Angioi M, Fay R, Nuee J et al. (2009) Extracellular cardiac matrix biomarkers in patients with acute myocardial infarction complicated by left ventricular dysfunction and heart failure: insights from the Eplerenone PostAcute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS) study. , Circulation; 119, 2471-9.
- 58.Ky B, French B, McCloskey K, Rame J E, McIntosh E et al. (2011) High-sensitivity ST2 for prediction of adverse outcomes in chronic heart failure. Circ Heart Fail;. 4, 180-7.
- 60.Felker G M, Fiuzat M, Thompson V, Shaw L K, Neely M L et al.Soluble ST2 in ambulatory patients with heart failure: association with functional capacity and long-term outcomes. Circ Heart Fail 2013;6:. 1172-9.
- 61.Sharma U C, Pokharel S, van Brakel TJ, van Berlo JH, Cleutjens J P et al. (2004) Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. , Circulation; 110, 3121-8.
- 62.Gruson D, Buglioni A, Burnett Jr JC. (2014) PTH: potential role in management of heart failure. , Clin Chim Acta; 433, 290-6.
- 63.Gruson D, Lepoutre T, Ketelslegers J M, Cumps J, Ahn S A et al. (2012) C-terminal FGF23 is a strong predictor of survival in systolic heart failure. , Peptides; 37, 258-62.
- 64.Bozkurt B. (2003) Use of biomarkers in the management of heart failure: are we there yet?. , Circulation; 107, 1231-3.
- 65.Kakkar R, Lee R T. (2008) The IL-33/ST2 pathway: therapeutic target and novel biomarker. , Nat Rev Drug Discov; 7, 827-40.
- 66.W J Paulus, Tschope C. (2013) A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. , J. Am. Coll. Cardiol;62: 263-271.
- 67.Glezeva J A Baugh. (2014) Role of inflammation in the pathogenesis of heart failure with preserved ejection fraction and its potential as a therapeutic target, Heart Fail. , Rev; 19, 681-694.
- 68.Akiyama E, Sugiyama S, Matsuzawa Y. (2012) Incremental prognostic significance of peripheral endothelial dysfunction in patients with heart failure with normal leftventricular ejection fraction. , J. Am. Coll. Cardiol; 60, 1778-1786.
- 69.Borlaug B A, Olson T P, Lam C S. (2010) Global cardiovascular reserve dysfunction in heart failure with preserved ejection fraction. , J. Am. Coll. Cardiol; 56, 845-854.
- 70.Borbely A, Falcao-Pires I, L van Heerebeek. (2009) Hypophosphorylation of the Stiff N2B titin isoform raises cardiomyocyte resting tension in failing human myocardium. , Circ. Res; 104, 780-786.
- 71.Takimoto E, Champion H C, Li M. (2005) Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. , Nat. Med; 11, 214-222.
- 72.Guazzi M, Vicenzi M, Arena R. (2012) Phosphodiesterase 5 inhibition with sildenafil reverses exercise oscillatory breathing in chronic heart failure: a long-term cardiopulmonary exercise testing placebo-controlled study. , Eur J Heart Fail;14: 82-90.
Cited by (1)
This article has been cited by 1 scholarly work according to:
Citing Articles:
Journal of Hypertension and Cardiology (2016) OpenAlex